
Poly(3-ethylglycolide): a well-defined polyester matching the hydrophilic hydrophobic balance of PLA†
 ‡ab
Nico
Ueberschaar,
d
Peter
Bellstedt,
a
Christine
Weber,
ab
Klaus D.
Jandt
*c
and
Ulrich S.
Schubert
*ab
‡ab
Nico
Ueberschaar,
d
Peter
Bellstedt,
a
Christine
Weber,
ab
Klaus D.
Jandt
*c
and
Ulrich S.
Schubert
*ab
Abstract
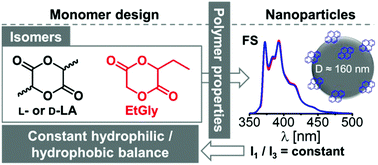
The ring opening (co)polymerization of 3-ethyl-1,4-dioxane-2,5-dione (3-ethylglycolide, EtGly) and enantiopure lactide using benzyl alcohol as initiator and the organobase 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene as catalyst yielded polyesters with predictable lactide and EtGly content between 5 and 20 mol%, molar masses around 11 000 g mol−1 and dispersities below 1.3. Due to the amorphous nature of the atactic poly(3-ethylglycolide) (PEtGly), dynamic scanning calorimetry revealed increasing glass transition and melting temperatures with an increasing lactide content of the statistical copolymers. Nanoparticles with a diameter of 160 nm and spherical shape were obtained from each polyester by applying the nanoprecipitation method, as confirmed by dynamic light scattering and scanning electron microscopy investigation. The constant hydrophilic to hydrophobic balance (HHB) of PLA and PEtGly was confirmed by fluorescence spectroscopy using pyrene loaded nanoparticles, confirming that a set of materials was obtained suitable to enlighten the effect of crystallinity on nanoparticle degradation.
 Please wait while we load your content...
Please wait while we load your content...

