
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A rapid, simple, high-performance liquid chromatography method for the clinical measurement of beta-lactam antibiotics in serum and interstitial fluid†
Alaa
Riezk
 *a,
Richard C.
Wilson
a,
Timothy M.
Rawson
a,
Vasin
Vasikasin
a,
Paul
Arkel
a,
Trevor J.
Ferris
b,
Lisa D.
Haigh
b,
Anthony E. G.
Cass
b and
Alison H.
Holmes
a
*a,
Richard C.
Wilson
a,
Timothy M.
Rawson
a,
Vasin
Vasikasin
a,
Paul
Arkel
a,
Trevor J.
Ferris
b,
Lisa D.
Haigh
b,
Anthony E. G.
Cass
b and
Alison H.
Holmes
a
aCentre for Antimicrobial Optimisation (CAMO), Imperial College London, Hammersmith Hospital, Du Cane Road, Acton, London, W12 0NN, UK. E-mail: a.riezk@imperial.ac.uk; alaa.riezk@yahoo.com; Tel: +44 (0)20 3313 2732
bDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College, London, UK
First published on 12th January 2023
Abstract
Background: enhanced methods of therapeutic drug monitoring are required to support the individualisation of antibiotic dosing based on pharmacokinetics (PK) parameters. PK studies can be hampered by limited total serum volume, especially in neonates, or by sensitivity in the case of critically ill patients. We aimed to develop a liquid chromatography–mass spectrometry (LC/MS) analysis of benzylpenicillin, phenoxymethylpenicillin and amoxicillin in single low volumes of human serum and interstitial fluid (ISF) samples, with an improved limit of detection (LOD) and limit of quantification (LOQ), compared with previously published assays. Methods: sample clean-up was performed by protein precipitation using acetonitrile. Reverse phase chromatography was performed using triple quadrupole LC/MS. The mobile phase consisted of 55% methanol in water + 0.1% formic acid, with a flow rate of 0.4 mL min−1. Antibiotics stability was assessed at different temperatures. Results: chromatographic separation was achieved within 3 minutes for all analytes. Three common penicillins can now be measured in a single low-volume blood and ISF sample (15 μL) for the first time. Validation has demonstrated the method to be linear over the range 0.0015–10 mg L−1, with an accuracy of 93–104% and high sensitivity, with LOD ≈ 0.003 mg L−1 and LOQ ≈ 0.01 mg L−1 for all three analytes, which is critical for use in dose optimisation/individualisation. All evaluated penicillins indicated good stability at room temperature over 4 h, at (4 °C) over 24 h and at −80 °C for 6 months. Conclusion: the developed method is simple, rapid, accurate and clinically applicable for the quantification of three penicillin classes.
Introduction
Since the discovery of penicillin, beta-lactams have remained the cornerstone of antibiotic management.1 Despite the subsequent discovery of many different classes of antibiotics, beta-lactams still account for more than a half of all prescribed antimicrobial agents in both the UK and worldwide.2,3 Four main classes of beta-lactam antibiotics are used in medical practice; penicillins, cephalosporins, monobactams, and carbapenems.Beta-lactam antibiotics remain popular due to their efficacy and safety profile. Regarding efficacy, they have a broad range of indications for the prevention and treatment of various types of infections, ranging from out-patient treatment of uncomplicated infections to the most serious infections in critically ill patients. Regarding safety, beta-lactams are typically considered to be among the safest classes of antibiotics available to clinicians.4,5
In the past few decades, antibiotic resistance has become an increasing challenge to global health.6 Several approaches have been utilised to address the challenge, including the use of greater doses and pairing beta-lactams with beta-lactamase enzyme inhibitors. Modified beta-lactams have also been developed that can bypass changes in organism cell membrane permeability, are less susceptible to enzymatic degradation or are less efficiently eliminated by the cell's efflux mechanisms.7 An additional intervention has been the development of therapeutic drug monitoring (TDM) to ensure that appropriate doses of antibiotics are prescribed to overcome the unpredictability and fluctuation of drug concentrations due to intra- and inter-individual variation in pharmacokinetics.8 Therefore, the measurement of beta-lactam concentrations in serum and interstitial fluid (ISF), as part of TDM, is becoming an important tool for optimising the treatment of infection to achieve successful outcomes whilst minimising the risk either of toxicity or the development of antimicrobial resistance.5,9 Therefore, the ex vivo measurement of beta-lactam concentrations is a crucial component of infection treatment optimisation through TDM required for immediate clinical application as well as in TDM research.10
Serum is commonly collected from patients by venepuncture and is routinely used to measure the concentration of various endogenous/exogenous substances in the blood compartment. ISF can be sampled using microdialysis techniques and measurements of antimicrobial concentrations are thought to be more representative of tissue concentration (i.e. the site of action).11
Many liquid chromatography–mass spectrometry (LC/MS) based bioanalytical methods have been described for beta-lactams. However, they have limitations; first, some are complicated, with multiple steps using different mobile phases for different beta-lactams,12,13 despite the requirement of simple, robust, and reliable results for TDM. Secondly, several studies reported a lower limit of detection (LOD) at above 0.1 mg L−1, much higher than the recommended therapeutic levels, which can be as low as 0.01 mg L−1.14 This means that such assays are not able to directly determine antibiotic target concentration attainment. Finally, most TDM studies measure total blood concentrations, although only the free antibiotic (non-protein bound) is pharmacologically effective and can diffuse from serum to the interstitial fluid and target infected tissue.15,16
Therefore, this study aimed to develop a fast, robust, sensitive, and clinically applicable triple quadrupole (TQ) LC/MS method to measure both the total and free concentrations of phenoxymethylpenicillin, benzylpenicillin and amoxicillin (Fig. 1) in both blood and interstitial fluid (ISF) samples obtained from human subjects. To our knowledge, this the first method to simultaneously determine phenoxymethylpenicillin, benzylpenicillin and amoxicillin in human ISF samples.
 | ||
| Fig. 1 Chemical structures of standard compounds. (A) Penicillin G, (B) penicillin V, (C) amoxicillin and (D) penicillin G-D7 (IS).27–30 | ||
Materials and methods
Chemicals and reagents
Benzylpenicillin sodium salt (penicillin G), phenoxymethylpenicillin potassium salt (penicillin V), amoxicillin trihydrate and benzylpenicillin-d7 N-ethylpiperidinium salt (used as an internal standard, IS) were purchased from Sigma (St. Louis, MO, USA), Fig. 1. Methanol, formic acid, water and acetonitrile in LC/MS grade were purchased from Sigma (St. Louis, MO, USA).Instrumentation
Chromatographic separation and mass-spectrometric detection were performed using a 1290 Infinity II LC equipped with a pump coupled to an Agilent Ultivo TQ LC/MS. Data were acquired and analysed using Agilent MassHunter software version 10.Sample and calibration preparation
Serum and ISF samples were obtained as a part of a clinical study evaluating the pharmacokinetics and pharmacodynamics of antibiotics in healthy volunteers. Serum and ISF samples were kept at −80 °C for further analysis. Before antibiotic administration, serum and ISF samples were collected from volunteers who had not taken any antibiotics before this first sample to make blank serum and ISF, which were subsequently spiked with nine different concentrations (0.0015–10 mg L−1) of each penicillin G, penicillin V and amoxicillin. The internal standard (penicillin G-D7) in acetonitrile (stored at −80 °C, prepared once every week) was added to each calibrator and sample at 0.15 mg L−1.A volume of 15 μL of biofluid (serum or ISF) was added to 60 μL of acetonitrile, vortexed, and allowed to equilibrate for 10 min. After that, precipitated proteins were separated by centrifugation for 5 min at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g.
000g.
Serum and ISF quality control specimens were prepared independently of standards at three concentration levels (Qc1; low; 0.0015 mg L−1, medium; Qc2; 0.123 mg L−1, and high; Qc3; 10 mg L−1). Aliquots of the Qc specimen were stored at −80 °C.
Chromatography conditions
Chromatography was performed on a reversed-phase analytical column (Poroshell 120 EC-C18, 2.1 × 50 mm; 1.9 micron, Agilent), using a mobile phase consisting of 55% methanol in water + 0.1% formic acid. Chromatographic separation was performed at a column temperature of 25 °C using a 0.4 mL min−1 flow rate, with an injection of 2 μL and a 6 min run time. Multiple reaction monitoring (MRM) in positive mode Electrospray Ionisation (ESI) was used for the detection of pencillins and both qualitative and quantitative data were collected. Table 1 shows MRM acquisition spectrometer parameters.| Analyte | Retention time (min) | Precursor/product ions (m/z) | Collision energy, CE (eV) |
|---|---|---|---|
| Penicillin G | 2.3 | 335.1/175.9; 160 | 12 |
| Penicillin V | 2.8 | 351.1/192; 159.8; 113.8 | 16 |
| Amoxicillin | 1 | 366.1/207.9; 113.9 | 8 |
| Penicillin G-D7, IS | 2.27 | 342.2/182.8; 160 | 12 |
Source parameters were optimised using the Agilent Source Optimizer program with the following parameters: gas temperature = 340 °C, gas flow = 9 L min−1, nebulizer = 40 psi, sheath gas flow = 250 °C and capillary voltage = 5000 V.
Stability experiments
Quality controls in serum and ISF samples (Qc1, Qc2, and Qc3) were stored at room temperature (RT) for 4 h and 24 h. After that, these sample were analysed and the data obtained at time = 0 h were compared to evaluate the short-term stability of the penicillin-containing serum samples. Similarly, the stability of samples in an autosampler was assessed at 4 °C. The long-term stability of analytes was assessed after freezing the serum samples at −80 °C for 6 months.The freeze–thaw stability of analytes was evaluated over three cycles within 3 days. In each cycle, Qc1, Qc2 and Qc3 were kept frozen at −80 °C and thawed at RT. When completely thawed, the samples were refrozen for 24 h at −80 °C.
The stability of penicillins was expressed as follows
| St (%) = C0/Ct × 100 |
Method validation and quality characterization
The analytical method was validated according to the International Conference on Harmonization guidelines.17 A triplicate run of calibration standards was performed every day for three consecutive days and triplicates of quality control samples (at low Qc1, medium Qc2, and high Qc3 concentrations) were carried out to evaluate the robustness of the developed method.Matrix effects
Standard solutions were prepared at appropriate concentrations depending on the expected drug level in three different sets. In the first set, standards of penicillin G, penicillin V, amoxicillin and IS were prepared in the mobile phase. A second set was made in blank serum samples or in blank ISF samples as a third set. The matrix effect was evaluated by comparing the peak area measurements obtained from the standard solutions.18| ME (%) = B/A × 100 |
A: the peak areas obtained from set 1 and B: the peak areas obtained from set 2 or 3. A value of 100% indicates that there is no absolute matrix effect.
Linearity
To assess linearity of the developed method, nine calibration standards were run for each analyte in triplicate over 3 consecutive days.Detection limit and quantification limit
The limit of detection (LOD) and limit of quantification (LOQ) were expressed based on the standard deviation of the response and the slope, with the following equations| LOD = 3.3 × &/s |
| LOQ = 10 × &/s |
& = the standard deviation of the response. s = the slope of the calibration curve.
Selectivity, precision and accuracy
Selectivity was assessed by analysing six different blank serum and ISF samples and confirmed by the absence of peaks at the respective retention times.The intra-day precision of the developed method was evaluated in terms of relative standard deviation (RSD) for the analysis of Qc1, Qc2 and Qc3 samples six times on the same day for intra-day precision. And inter-day precision was determined by the analysis of triplicate QC samples on five consecutive days. The accuracy of the method was determined by the percentage agreement between the measured and spiked concentrations using the following formula:
| Accuracy percentage = measured/known spiked × 100 |
Carry-over
The carry-over of analytes and IS was assessed by analysing blank samples after the highest standard sample concentration 10 mg L−1 was reached. Then, standard samples at a lower limit of quantification (0.0015 mg L−1) were also analysed after the blank samples. The experiment was repeated three times. Carry-over in the blank samples should not be greater than 20% of the analyte response at the lower limit of quantification and 5% of the response for the IS.17Pharmacokinetic study
A healthy volunteer from an ongoing clinical trial is included in this study as an example of the application of this method to clinical samples. The volunteer received penicillin G 1,200 mg intravenously over 5 minutes. A separate cannula for blood sampling was inserted in a large vein in the antecubital fossa. A microdialysis catheter was inserted subcutaneously in the forearm. The blood and ISF samples were taken via a cannula and by microdialysis, respectively. The study is approved by the London Harrow Research Ethics Committee (19/LO/0219) and registered on https://www.ClinicalTrials.gov (NCT04053140). And participants gave informed written consent.Serum protein binding
A method to quantify penicillin G fractions bound and unbound to serum proteins was developed by using a Centrifree® Ultrafiltration Device (Merck Millipore). The influence of temperature, pH and relative centrifugation forces (RCFs) on the protein binding was investigated. Accordingly, the optimised conditions for a higher recover were when unbound penicillin G was quantified by pipetting 0.7 mL of serum at a pH of 7.4 using 0.1 M sodium phosphate buffer, and centrifuging at 1500 g for 10 minutes at 25 °C. The filtrate was then analysed by LC/MS as described before.The free fraction of drug was calculated as follows:
| Free drug (%) = (Cfree/Ctotal) × 100. |
C free: concentration of the free drug. Ctotal: concentration of the total drug.
Results and discussion
The aim of the project was to develop a reliable and rapid analytical method for the quantification of penicillin G, penicillin V and amoxicillin in both ISF and human serum to provide reliable pharmacokinetic data after drug administration, including towards the end of a dosage interval when serum concentrations are low.Different combinations and gradients of mobile phases (consisting of water and acetonitrile or methanol) and reverse-phase uHPLC columns were assessed. Other parameters, such as using different columns (Poroshell 120 EC-C18; 2.1 × 50 mm; 1.9 micron (Agilent), Polaris 5 C18-A; 5 micron (Agilent) and Kinetex; 50 × 4.6; 5 microns (Phenomenex), oven temperature (25 °C and 40 °C), injection volume (1, 2 and 4 μL) and mobile phases (using different ratios of methanol, acetonitrile and formic acid), were evaluated in order to obtain the simultaneous measurements of the concentrations of the three analytes with a short retention time and high response.
Optimised conditions consisted of a mobile phase of 55% methanol in water + 0.1% formic acid, Poroshell 120 EC-C18, 25 °C column temperature, 2 μL injection and a flow rate of 0.4 mL min−1. This achieved the highest MS response and produced sharp peaks at retention times 2.3, 2.8, 1, and 2.27 min for penicillin G, penicillin V, amoxicillin and IS respectively (Fig. S1†), and the observed chromatographic data were used in quantification.
Deproteinisation processes were analysed using different volumes of acetonitrile and samples to choose the sample pre-treatment process that leads to a greater recovery. The selected process was 15 μL of sample and 60 μL of acetonitrile with recovery greater than 98%. Several steps of optimisation led to a developed method with one mobile phase, short retention time, small sample volume and a greatly improved LOD and LOQ.
The relative detector response (peak area), when plotted vs. the injected relative concentration to IS, was found to be linear over the concentration range 0.0015–10 mg L−1 with a correlation coefficient (R2 = 1, Table 2 and Fig. S2† shows the calibration parameters for the three analytes along with the LOD and LOQ.
| Analyte | Slope | Intercept | R 2 | LOD (mg L−1) | LOQ (mg L−1) |
|---|---|---|---|---|---|
| a R 2, coefficient of determination. | |||||
| Penicillin G | 0.7539 | 0.0002 | 1 | 0.003 | 0.01 |
| Penicillin V | 0.6632 | 0.0002 | 1 | 0.003 | 0.01 |
| Amoxicillin | 0.5858 | 0.0002 | 1 | 0.003 | 0.01 |
In this study, we aimed to address two main concerns: first of all the demand for relatively large sample volumes to assess drug levels in blood, which impedes meaningful paediatric clinical trials, because of ethical limitations on children regarding blood sampling (such as discomfort, fear and restricted blood volumes). Secondly, penicillins exhibit mainly time-dependent bactericidal action; the longer the concentration is above the MIC, the greater the bactericidal effect.19 TDM can improve pharmacological target attainment, with potential to improve patient outcomes.19 Current assays often fail to measure around organisms (such as streptococcus pyogenes and staphylococcus aureus) with MIC less than 0.1 mg L−1.20,21
This study was able to report the measurement of penicillin G, penicillin V and amoxicillin in a single LC/MS assay using a low sample volume of 15 μL, with the ability to detect and quantify at low concentrations, which is much more sensitive than other published data (10 to 33 times) which reported a LOD and LOQ of around 0.1 mg L−1.22–24
Matrix effect recovery, accuracy and carry-over
The matrix recovery was 97–102% for penicillin G, 98–103% for penicillin v, 97–102% for amoxicillin in both serum and ISF samples and 99–103% for IS, indicating that the matrix effect did not significantly interfere with the calibration, as the internal standard corrected for any matrix effects.25Accuracy and precision were tested at three different concentrations (Qc1, Qc2 and Qc3, Table 3): in intra-day and inter-day runs accuracies for all analytes in both blood and ISF samples ranged from 93% to 104%, while precision for all analytes in both blood and ISF samples in intra-day and inter-day runs ranged from 0.5 to 1.8%. Meanwhile, Kepper et al. 2018 reported an inter- and intra-precision of 2–5.7% for benzylpenicillin and of 3.8–6.1% for amoxicillin in serum samples.12 Khuroo et al. 2008 showed that in her LC/MS method for amoxicillin the intra-day precision is from 1.3% to 8.8% and the inter-day precision is from 1.8% to 6.2%.26
| Spiking level (mg L−1) | RSD (%) | Accuracy (%) | |||
|---|---|---|---|---|---|
| Serum | ISF | Serum | ISF | ||
| Intra-day | |||||
| Qc1 | 10 | 0.8 | 1 | 102 | 101 |
| Qc2 | 0.123 | 1.1 | 1.1 | 99 | 100 |
| Qc3 | 0.0015 | 1.6 | 1.5 | 93 | 104 |
|
|||||
| Inter-day | |||||
| Qc1 | 10 | 1.7 | 1.8 | 99 | 103 |
| Qc2 | 0.123 | 0.5 | 1.2 | 99 | 102 |
| Qc3 | 0.0015 | 1.8 | 1.5 | 95 | 93 |
Carry-over for all analytes was considered acceptable for all analytes and IS with less than 3%.
Stability study
For penicillins, long-term stability studies showed no significant degradation in Qc samples (in both serum and ISF) stored at −80 °C for 6 months. All penicillins in serum and ISF samples remained stable within the range of 97 to 104% when kept at room temperature (RT) ((23 ± 2 °C) for 4 h (Fig. 2).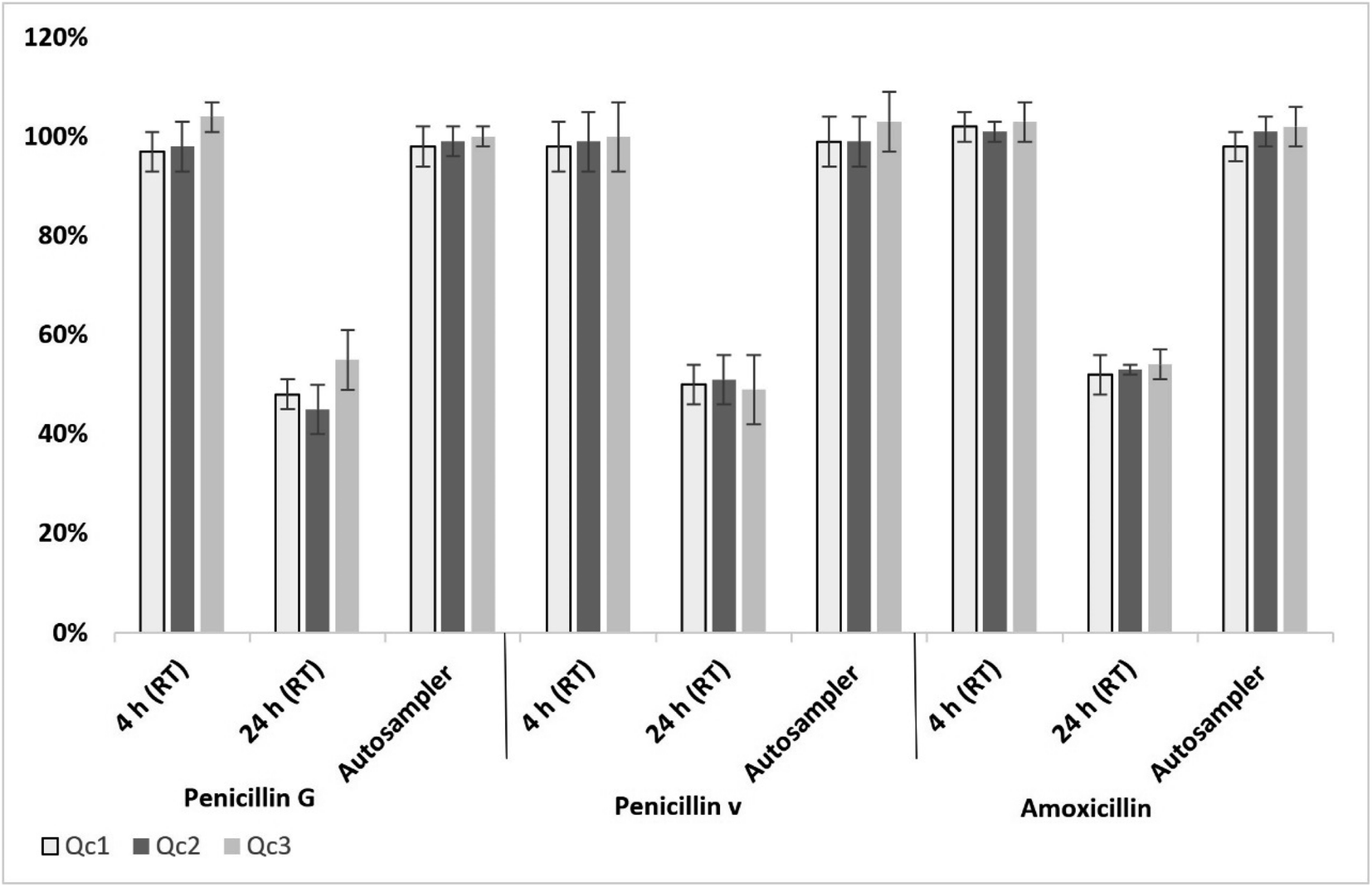 | ||
| Fig. 2 Average stability of penicillins in serum over 4 h and 24 h at RT and over 24 h in the autosampler (n = 5); error bars represent standard deviation. | ||
After 24 h storage at RT of penicillin-spiked serum or ISF (Qc1, Qc2 and Qc3), a significant degradation was observed with a detection range of 45–55% of the analytes (Fig. 2). All penicillins in extracted serum or ISF samples indicated a good stability when kept in an autosampler (4 °C) over 24 h, remaining in the range of 98 to 103% from the original concentration (Fig. 2).
No penicillin degradation was observed when subjected to three freeze–thaw cycles (ISF or serum).
Application to clinical samples
The method has been used in a clinical study measuring total penicillin G and free serum and ISF concentrations in healthy volunteers. The serum and ISF concentrations of a participant are shown in Fig. 3. These measurements can be fitted to a standard pharmacokinetic human model.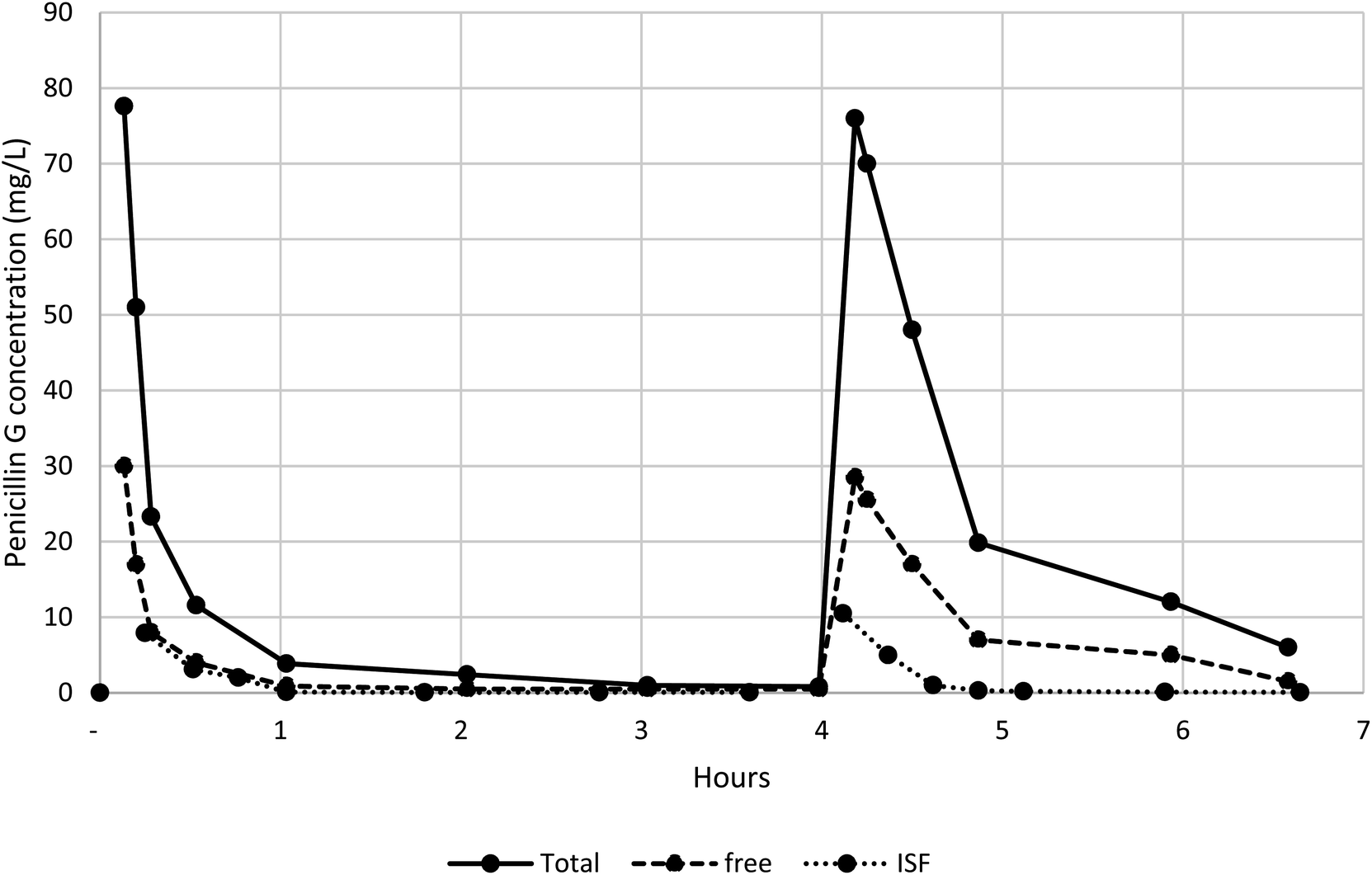 | ||
| Fig. 3 Penicillin G total and free serum and ISF concentrations at various times in a participant. | ||
Conclusion
The method presented here has been successfully used in a clinical study with healthy volunteers measuring penicillin total and free serum concentrations and ISF concentrations. It is rapid, accurate, and robust, and has a low LOD and LOQ and a simple sample preparation. This may help in optimising treatment to improve patient outcomes, whilst minimising the risk of development of antibiotic resistance.Institutional review board statement
The study is approved by the London Harrow Research Ethics Committee (19/LO/0219) and is registered on https://www.ClinicalTrials.gov (NCT04053140).Data availability
Data and materials are available from the authors on reasonable request.Author contributions
Conceptualization: AR, RCW, TMR, VV, AEGC, AEGC and AHH. Formal analysis: AR. Investigation: AR, RCW, TMR, VV, AEGC and AHH. Methodology: AR. Project administration: TMR and AHH. Validation: AR. Visualization: AR, RCW, TMR, VV, PA, TJF, LDH, AEGC and AHH. Writing – original draft: AR. Writing – review & editing: AR, RCW, TMR, VV, PA,TJF, LDH, AEGC and AHH.Conflicts of interest
Timothy M. Rawson has received honoraria from Sandoz (2020), bioMérieux (2021/2022) and Roche Diagnostics (2021). All other authors have no potential conflicts of interest to declare.Acknowledgements
The Department of Health and Social Care funded the Centre for Antimicrobial Optimisation (CAMO), Imperial College London provided state-of-the-art research facilities and consolidated multidisciplinary academic excellence and clinical expertise, and Imperial's NIHR/Wellcome funded the Clinical Research Facility (CRF) and partnerships with the NHS to support and deliver innovative research on antimicrobial optimisation and precision prescribing. We would like to acknowledge: (1) the Department of Health & Social Care that funded the Centre for Antimicrobial Optimisation (CAMO) at Imperial College London; (2) the National Institute for Health and Care Research Health Protection Research Unit (NIHR HPRU) in Healthcare Associated Infections and Antimicrobial Resistance at Imperial College London; (3) The Agilent Measurement Suite at Imperial College London. The authors are grateful to Kerri Hill-Cawthorne and Gurjeeta Bhangal, from CAMO, for assistance and support.References
- C. L. Tooke, P. Hinchliffe, E. C. Bragginton, C. K. Colenso, V. H. A. Hirvonen, Y. Takebayashi and J. Spencer, J. Mol. Biol., 2019, 431, 3472–3500 CrossRef CAS PubMed.
- WHO report on surveillance of antibiotic consumption, https://www.who.int/publications/i/item/who-report-on-surveillance-of-antibiotic-consumption.
- F. C. K. Dolk, K. B. Pouwels, D. R. M. Smith, J. V. Robotham and T. Smieszek, J. Antimicrob. Chemother., 2018, 73, ii2–ii10 CrossRef CAS PubMed.
- R. C. Owens Jr, Crit. Care, 2008, 12(4), S3 CrossRef PubMed.
- A. Rodríguez-Gascón, M. Á. Solinís and A. Isla, Pharmaceutics, 2021, 13, 833 CrossRef PubMed.
- A. H. Holmes, L. S. Moore, A. Sundsfjord, M. Steinbakk, S. Regmi, A. Karkey, P. J. Guerin and L. J. Piddock, Lancet, 2016, 387, 176–187 CrossRef CAS PubMed.
- A. J. Kunz Coyne, A. El Ghali, D. Holger, N. Rebold and M. J. Rybak, Infect. Dis. Ther., 2022, 11, 661–682 CrossRef PubMed.
- J. Gonçalves-Pereira and P. Póvoa, Crit. Care, 2011, 15, R206 CrossRef PubMed.
- R. Guilhaumou, S. Benaboud, Y. Bennis, C. Dahyot-Fizelier, E. Dailly, P. Gandia, S. Goutelle, S. Lefeuvre, N. Mongardon, C. Roger, J. Scala-Bertola, F. Lemaitre and M. Garnier, Crit. Care, 2019, 23, 104 CrossRef PubMed.
- J. A. Roberts, M. Ulldemolins, M. S. Roberts, B. McWhinney, J. Ungerer, D. L. Paterson and J. Lipman, Int. J. Antimicrob. Agents, 2010, 36, 332–339 CrossRef CAS PubMed.
- M. Brunner, H. Stabeta, J. G. Möller, C. Schrolnberger, B. Erovic, U. Hollenstein, M. Zeitlinger, H. G. Eichler and M. Müller, Antimicrob. Agents Chemother., 2002, 46, 3724–3730 CrossRef CAS PubMed.
- K. Kipper, C. I. S. Barker, J. F. Standing, M. Sharland and A. Johnston, Antimicrob. Agents Chemother., 2018, 62, e01540–17 CrossRef PubMed.
- B. C. McWhinney, S. C. Wallis, T. Hillister, J. A. Roberts, J. Lipman and J. P. Ungerer, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2039–2043 CrossRef CAS PubMed.
- M. H. Abdul-Aziz, K. Brady, M. O. Cotta and J. A. Roberts, Ther. Drug Monit., 2022, 44, 19–31 CrossRef CAS PubMed.
- J. Beer, C. C. Wagner and M. Zeitlinger, AAPS J., 2009, 11, 1–12 CrossRef CAS PubMed.
- K. K. Hansen, F. Nielsen, T. B. Stage, U. Jørgensen, O. Skov and L. E. Rasmussen, Br. J. Clin. Pharmacol., 2018, 84, 533–541 CrossRef CAS PubMed.
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, ICH Harmonised Guideline, Validation of Analytical Procedures, Q2(R2), https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-q2r2-validation-analytical-procedures-step-2b_en.pdf.
- J. C. Van De Steene and W. E. Lambert, J. Am. Soc. Mass Spectrom., 2008, 19, 713–718 CrossRef CAS PubMed.
- T. Luxton, N. King, C. Wälti, L. Jeuken and J. Sandoe, J. Antimicrob. Chemother., 2022, 77, 1532–1541 CrossRef CAS PubMed.
- M. Camara, A. Dieng and C. S. Boye, Microbiol. Insights, 2013, 6, 71–75 Search PubMed.
- Y.-A. Que and P. Moreillon, in Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases, ed. J. E. Bennett, R. Dolin and M. J. Blaser and W. B. Saunders, Philadelphia, 8th edn, 2015, pp. 2237–2271.e2235, DOI: DOI:10.1016/B978-1-4557-4801-3.00196-X.
- L. A. Decosterd, T. Mercier, B. Ternon, S. Cruchon, N. Guignard, S. Lahrichi, B. Pesse, B. Rochat, R. Burger, F. Lamoth, J. L. Pagani, P. Eggimann, C. Csajka, E. Choong, T. Buclin, N. Widmer, P. André and O. Marchetti, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2020, 1157, 122160 CrossRef CAS PubMed.
- S. Pingale, M. Badgujar, K. Mangaonkar and N. Mastorakis, WSEAS Trans. Biol. Biomed., 2012, 1, 1–13 Search PubMed.
- T. Ohmori, A. Suzuki, T. Niwa, H. Ushikoshi, K. Shirai, S. Yoshida, S. Ogura and Y. Itoh, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1038–1042 CrossRef CAS PubMed.
- A. De Nicolò, M. Cantu and A. D’Avolio, Bioanalysis, 2017, 9, 1093–1105 CrossRef PubMed.
- A. H. Khuroo, T. Monif, P. R. P. Verma and S. Gurule, J. Chromatogr. Sci., 2008, 46, 854–861 CAS.
- National Center for Biotechnology Information, PubChem Compound Summary for CID 5904, Penicillin g, 2022, https://pubchem.ncbi.nlm.nih.gov/compound/Penicillin-g Search PubMed.
- National Center for Biotechnology Information, PubChem Compound Summary for CID 6869, Penicillin v, 2022, https://pubchem.ncbi.nlm.nih.gov/compound/Penicillin-v Search PubMed.
- National Center for Biotechnology Information, PubChem Compound Summary for CID 33613, Amoxicillin, 2022, https://pubchem.ncbi.nlm.nih.gov/compound/Amoxicillin Search PubMed.
- National Center for Biotechnology Information, PubChem Compound Summary for CID 3084757, Benzylpenicilline-d7 N-Ethylpiperidinium Salt, 2022, https://pubchem.ncbi.nlm.nih.gov/compound/Benzylpenicilline-d7-N-ethylpiperidinium-salt Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ay01276f |
| This journal is © The Royal Society of Chemistry 2023 |