
Arsenite-induced apoptosis can be attenuated via depletion of mTOR activity to restore autophagy†
Chien-Wei
Wu
,
Pei-Jung
Lin
,
Jia-Shiuan
Tsai
,
Chih-Ying
Lin
and
Lih-Yuan
Lin
 *
*
Institute of Molecular and Cellular Biology and Department of Life Science, National Tsing Hua University, Hsinchu, Taiwan. E-mail: lylin@life.nthu.edu.tw; Tel: +886-3-5742693
First published on 30th October 2018
Abstract
Arsenic and its compounds are toxic environmental pollutants and known carcinogens. We investigated here the mechanism of arsenite-induced damage in renal cells. Treating human embryonic kidney cells (HEK293) with sodium arsenite reduces cell viability in a dose- and time-dependent manner. The decline of cell viability is due to apoptotic death since arsenite treatment reduces Akt activity and the Bcl2 level but increases caspase 3 activity and the cytochrome c level. These effects can be reverted by the addition of an apoptosis inhibitor. PTEN, the upstream negative regulator of Akt activity, was also reduced with arsenite treatment. Noticeably, PTEN markedly increased in the insoluble fraction of the cells, suggesting a cell failure in removing the damaged proteins. Arsenite treatment activates a variety of signaling factors. Among them, ERK and JNK are associated with autophagy via regulating the levels of LC3 and p62. With arsenite administration, the LC3 and p62 levels increased. However, lysosomal activity was decreased and led to the decline of autophagic activity. The addition of rapamycin, the mTOR inhibitor, activated the autophagic pathway that accelerated the removal of damaged proteins. The recovery of autophagy increased the viability of arsenite-treated cells. Similar to rapamycin treatment, the knockdown of mTOR expression also enhanced the viability of arsenite-treated cells. Both rapamycin treatment and mTOR knockdown enhanced ERK activity further, but reduced JNK activity and the p62 level in arsenite-treated cells. Lysosomal activity increased with the depletion of mTOR, indicating an increase of autophagic activity. These results reveal the critical role of mTOR in regulating the cell fate of arsenite-exposed renal cells.
Introduction
Arsenic compounds are widely distributed in the environment. They are commonly used in agriculture, industry and medication. Due to the massive application of these chemicals, serious arsenic contamination occurred in many countries. The toxicities of arsenic compounds are well documented. Environmental or occupational exposures to arsenic compounds are associated with the manifestations of skin lesions, diabetes, cardiovascular disease and various types of cancers.1 Arsenic exposure is known to increase the risk of skin, lung, liver, kidney, prostate, and urinary bladder cancers.2 Two common mechanisms are recognized for arsenic-induced genotoxicity: arsenic exposure causes oxidative stress via the formation of reactive oxygen species (ROS) and it inhibits DNA repairs.3 ROS production upon arsenite induction has multiple effects depending on the cell types. It can cause apoptosis, necrosis, autophagy, cell cycle arrest, DNA strand breaks, and gene mutation.4–6 The inhibition of DNA repair promotes the DNA-damaged cells toward carcinogenesis.Apoptosis can act as a defense mechanism to reduce tumor development.7 The occurrence of apoptosis can be initiated through an exogenous (death receptors) or endogenous (mitochondria) pathway. The endogenous apoptotic machinery is regulated mainly by pro- and anti-apoptotic members of the Bcl2 family.7 Under stress stimulation, the anti-apoptotic activity of Bcl2 is inhibited. The pro-apoptotic Bax proteins then move to the mitochondria.8 The Bax proteins oligomerize and insert into the outer membrane of the mitochondria to form channels for the release of cytochrome c from the mitochondria.9 Cytochrome c then binds with apoptotic protease activating factor 1 (Apaf-1) and recruits caspase 9 to form an apoptosome which further activates caspase 3 and leads cells to apoptotic death.10
Autophagy plays a dual role in tumor development. It helps tumor growth, metabolism, and survival via nutrient recycling.11,12 Conversely, autophagy can reduce tumor initiation by suppressing cell damage through its quality control function.13 Autophagy can be activated by various stresses. It serves as a primary response to remove damaged proteins, DNA or organelles. However, with sustained stress, cells can no longer remove the accumulated damage and instead initiate the death response.14
Autophagy can be characterized as macroautophagy, microautophagy and chaperone-mediated autophagy. Among them, macroautophagy is frequently reported. In the presence of damaged DNA, organelles or aggregated proteins, an ubiquitin-binding protein, p62, targets the damaged materials. Autophagosomes with a bi-layer membrane structure are formed to engulf the damaged materials. Microtubule-associated protein 1A/1B light chain 3 (LC3) binds on the membrane with other proteins to accelerate the formation of autophagosomes. The matured autophagosomes then merge with lysosomes to form autolysosomes with a single-layer membrane structure. LC3 dissociates from the structure and enzymes in the lysosome digest the damaged materials.15–17 LC3 and p62 are thus usually regarded as markers of autophagy.
Apoptosis and autophagy can be regulated by the mammalian target of rapamycin (mTOR). The role of mTOR can be either pro- or anti-apoptotic. Activation of S6K, a downstream target of mTOR, inhibits the activity of pro-apoptotic factor BAD and thus reduces apoptosis.18,19 Conversely, mTOR can move into the nucleus to phosphorylate p53 which enhances the transcription of Bax.20 The increase of this pro-apoptotic factor stimulates apoptosis. mTOR also regulates autophagy. The activation of mTOR phosphorylates ULK1 and inhibits the initiation of autophagy.21,22 mTOR also targets the vps34 protein complex to block the formation of autophagosomes.21,23 Therefore, an increase in mTOR activity inhibits autophagy.
Reportedly, arsenic treatment activates a variety of signaling pathways that modulate the cell fate.24–26 However, the underlining mechanism to explain the effect of arsenic on cell death needs further clarification. Apoptosis is generally recognized as the major type of cell damage with arsenic exposure. Recent studies also indicate the participation of autophagy in arsenic-insulted cells.25,27 We therefore investigated whether the cross-interaction of apoptosis and autophagy occurs in arsenic-treated cells. Additionally, whether the arsenic-induced cell damage can be attenuated by altering the related pathways remains to be explored. In this study, we treated human embryonic kidney 293 (HEK293) cells with sodium arsenite. We found that arsenite treatment damaged cellular components and activated autophagy. However, the autophagy was blocked and caused the accumulation of damaged materials in cells. Apoptosis has thus occurred. Noticeably, the blockade of autophagy can be released by depleting the mTOR activity. The autophagy process attenuates the arsenite-treated cells from apoptotic death.
Materials and methods
Cell cultures and chemicals
Human embryonic kidney 293 (HEK293, ATCC® CRL1573™) cells were cultured at 37 °C in Dulbecco's modified Eagle's medium (DMEM) with 10% heat-inactivated fetal bovine serum (FBS), 0.015% glutamine, 0.37% sodium bicarbonate, 100 units per ml penicillin, and 100 mg ml−1 streptomycin in 5% CO2, 95% air and 100% humidity. Cell culture reagents were purchased from Invitrogen/Gibco. Lipofectamine™ RNAiMAX was purchased from Invitrogen. p-Nitrophenyl phosphate (pNPP) and benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (z-VAD-fmk) were purchased from Sigma-Aldrich. Necrostatin-1 and the caspase substrate Ac-DEVD-AFC were from Calbiochem. Anti-GAPDH antibodies were purchased from Gene Tex. Anti-p-mTOR, anti-mTOR, anti-p-Akt (S473), anti-Akt, anti-PTEN, anti-LC3B, anti-p-JNK, anti-JNK, anti-p-ERK, anti-ERK, anti-p-p38 and anti-p38 antibodies were from Cell Signaling Technology. Anti-cytochrome c and anti-Bcl2 antibodies were from Santa Cruz Biotechnology. Anti-p62 antibodies were purchased from BD Transduction Laboratories™. Protein markers were from SMOBIO. Other chemicals were purchased from Sigma-Aldrich unless specified.Cell viability assay
HEK293 cells were seeded in 96-well plates at a density of 1.5 × 104 cells per well and treated with 0, 10, 20, 40 and 60 μM of sodium arsenite for 24 h. MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; 0.4 mg ml−1] was added to the cells 4 h before harvesting. Cells were first washed with phosphate-buffered saline (PBS) and then 200 μl DMSO was added into each well. The absorbance at 550 nm was recorded with a ThermoMax microplate reader (Molecular Devices). For time course studies, cells were treated with 20 μM of sodium arsenite for 0, 3, 6, 12 and 24 h before the MTT assay was conducted.Immunoblotting analysis
Cells were harvested, centrifuged (1200g for 5 min) and then resuspended in 10 cell volumes of lysis buffer (20 mM HEPES, pH 7.9, 400 mM NaCl, 0.5 mM PMSF, 50 mM NaF, 0.5 mM Na3VO4, 2 μg ml−1 aprotinin, 5 μg ml−1 leupeptin, 1 μg ml−1 pepstatin, and 0.5% NP-40) after treatments. The cells were incubated on ice and lysed by vortexing vigorously four times for 15 s every 10 min. The cell lysate was obtained by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 min at 4 °C. The supernatants (soluble fraction, SF) or the pellets (insoluble fraction, InF) were collected. Five cell volumes of a loading dye (0.125 M Tris, pH 6.8, 0.4% SDS, 20% glycerol, and 0.04% bromophenol blue) were added to the InF and then boiled for 10 min. The whole cell proteins were obtained by spinning down the cells, adding 10 cell volumes of the loading dye to the cell pellet and boiling for 10 min. An equal volume of 2× loading dye was added to the SF samples before loading onto the gel. The samples were separated electrophoretically on a 10% SDS-polyacrylamide gel (5% stacking gel). Separated proteins were transferred onto a PVDF membrane (GE Healthcare) with a transfer cell (Bio-Rad). The membrane was pre-hybridized in TBST buffer (150 mM NaCl, 10 mM Tris-Cl, pH 8.0, 0.1% Tween-20) with 5% skimmed milk for at least 1 h, and then changed to TBST buffer with 5% skimmed milk and primary antibodies at 4 °C overnight. The membrane was washed three times with TBST buffer for 15 min each, and then immersed in 5% skimmed milk containing horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. After washing three times with TBST buffer, the membrane was rinsed once with TBS buffer (TBST buffer without Tween-20), and developed using an ECL system (Amersham). Visualization was achieved by using an ImageQuant™ LAS 4000 mini imager (GE Healthcare). The intensities of the protein bands were quantified using the UN-SCAN-IT gel analysis software (version 6).
000g for 20 min at 4 °C. The supernatants (soluble fraction, SF) or the pellets (insoluble fraction, InF) were collected. Five cell volumes of a loading dye (0.125 M Tris, pH 6.8, 0.4% SDS, 20% glycerol, and 0.04% bromophenol blue) were added to the InF and then boiled for 10 min. The whole cell proteins were obtained by spinning down the cells, adding 10 cell volumes of the loading dye to the cell pellet and boiling for 10 min. An equal volume of 2× loading dye was added to the SF samples before loading onto the gel. The samples were separated electrophoretically on a 10% SDS-polyacrylamide gel (5% stacking gel). Separated proteins were transferred onto a PVDF membrane (GE Healthcare) with a transfer cell (Bio-Rad). The membrane was pre-hybridized in TBST buffer (150 mM NaCl, 10 mM Tris-Cl, pH 8.0, 0.1% Tween-20) with 5% skimmed milk for at least 1 h, and then changed to TBST buffer with 5% skimmed milk and primary antibodies at 4 °C overnight. The membrane was washed three times with TBST buffer for 15 min each, and then immersed in 5% skimmed milk containing horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. After washing three times with TBST buffer, the membrane was rinsed once with TBS buffer (TBST buffer without Tween-20), and developed using an ECL system (Amersham). Visualization was achieved by using an ImageQuant™ LAS 4000 mini imager (GE Healthcare). The intensities of the protein bands were quantified using the UN-SCAN-IT gel analysis software (version 6).
Caspase 3 activity assay
Cells were harvested and lysed in 10 cell volumes of extraction buffer (1% Triton X-100, 1% NP-40, 2 μg ml−1 aprotinin, 5 μg ml−1 leupeptin, and 2 mM PMSF) and incubated on ice for 10 min. After centrifugation at 12000g and 4 °C for 30 min, the supernatants were collected and protein concentrations were determined with a protein assay kit (Bio-Rad). Fifty μg of proteins were incubated at 37 °C in 100 μl reaction buffer (10 mM HEPES, pH 7.9, 2 mM EDTA, 10 mM KCl, 1.5 mM MgCl2, 10 mM DTT) with 50 μM caspase substrate Ac-DEVD-AFC for 1 h. The fluorescence from AFC was detected at excitation and emission wavelengths of 405 and 510 nm, respectively, with a Wallac 1420 Multilabel Counter Victor2 (Perkin Elmer).
Acid phosphatase activity assay
Whole cell lysates were prepared as that for immunoblotting except that NaF was omitted from the lysis buffer. Fifty μg of protein extracts were mixed with 50 μg of pNPP in 100 μl citrate buffer (40 mM citric acid monohydrate and 60 mM trisodium citrate dihydrate) and incubated at 37 °C for 20 min. The reaction was terminated by adding 50 μl of 0.5 N NaOH at room temperature. Samples (100 μl) were transferred to the 96-well plate. The reaction product, p-nitrophenol, was detected at a 405 nm wavelength using a ThermoMax microplate reader (Molecular Devices).RNA interference
HEK293 cells (5 × 105) were seeded in 6-well plates with 2.5 ml medium without antibiotics for 24 h (approximately 50% confluence). Transfections were conducted using siRNA and Lipofectamine™ RNAiMAX following the manufacturer's instructions. The final concentration of the siRNA in the well was 20 nM. Transfection of the siRNA was performed at 37 °C for 48 h.Statistical analysis
All experiments were repeated three times independently and the means ± standard deviations of the samples were calculated. Significant differences between two treatments were analyzed by two-tailed Student's t test.Results
Cytotoxicity effect of sodium arsenite on HEK293 cells
To investigate the mechanism of arsenite-induced cytotoxicity, HEK293 cells were treated with 0–60 μM of sodium arsenite for 24 h. Cell viability was analyzed. Fig. 1A shows that cell viability decreased in a dose-dependent manner. A time course study was subsequently conducted by treating cells with 20 μM arsenite for 0–24 h. The results demonstrate that cell viability reduced in a time-dependent manner (Fig. 1B). Cells treated with 20 μM arsenite for 24 h resulted in a 50% viability. This condition was used in the following studies unless specified.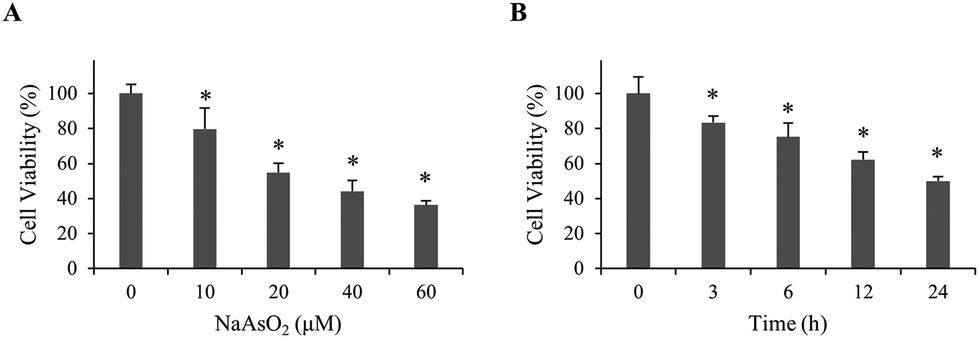 | ||
| Fig. 1 Viability of renal cells with NaAsO2 treatment. (A) HEK293 cells were treated with various concentrations of NaAsO2 for 24 h and cell viability was determined by the MTT assay. (B) HEK293 cells were treated with 20 μM of NaAsO2 for various time intervals. Cell viability was determined by the MTT assay. Each value represents a mean ± standard deviation of three independent samples. Asterisk indicates the significant difference as compared to that of the untreated cells. *p < 0.05. | ||
Arsenite treatment induces apoptosis in HEK293 cells
Different types of cell deaths have been reported with arsenite treatment.25,28 In order to identify the mechanism of cell death, an inhibitor for apoptosis (z-VAD-fmk), necrosis (necrostatin-1) or autophagy (NH4Cl) was added with arsenite to cells. Cell viability was determined. The results show that z-VAD-fmk significantly increased the cell viability (Fig. 2A). This finding suggests that apoptosis occurred with arsenite treatment.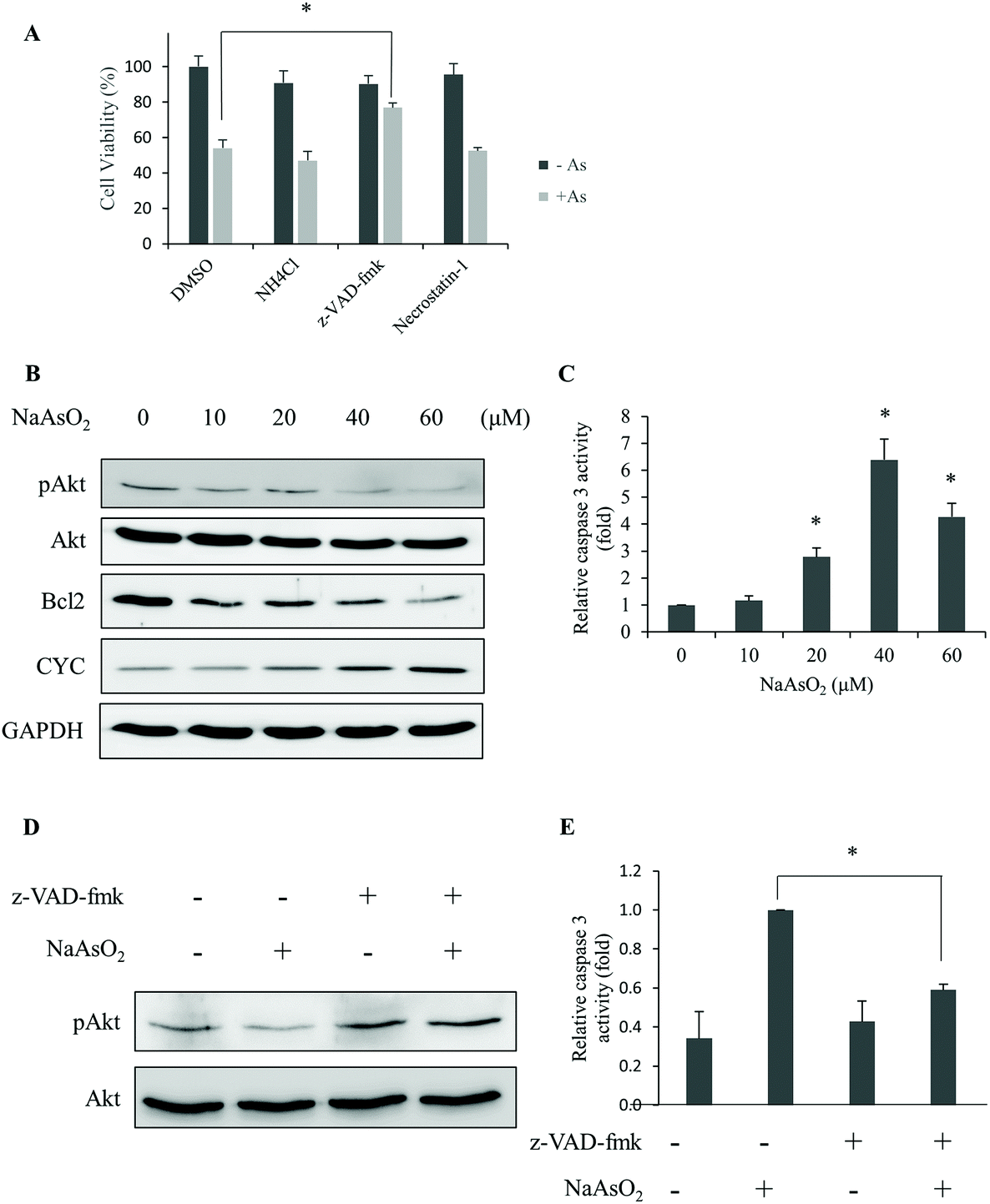 | ||
| Fig. 2 Arsenite treatment causes apoptotic death in renal cells. (A) HEK293 cells were treated with 20 μM NaAsO2 for 24 h in the presence of the inhibitor for autophagy (NH4Cl), apoptosis (z-VAD-fmk) or necrosis (Necrostatin-1). Cell viability was determined. The black and gray bars indicate cells without and with arsenite treatment, respectively. (B) Phosphorylated Akt (pAkt), Bcl2, and cytochrome c (CYC) derived from cells treated with various concentrations of NaAsO2 for 24 h were analyzed by immunoblotting. GAPDH was used as a loading control. (C) HEK293 cells were treated with various concentrations of NaAsO2 for 24 h and the caspase 3 activities were determined. (D) HEK293 cells were treated with and without arsenite in the presence or absence of 20 μM z-VAD-fmk. The Akt and pAkt levels in the samples were visualized with immunoblotting. (E) The caspase 3 activity of samples in (D) was determined. Results in (D) and (E) were normalized to those of samples without NaAsO2 treatment and reported as fold increase. Values in (A), (C), and (E) represent a mean ± standard deviation of three independent samples. Asterisks indicate significant differences as compared to that of the untreated cells (A and C) or the paired samples (E). *p < 0.05. | ||
Reportedly, Akt activity is affected by arsenic treatment with subsequent cell apoptosis.26 Hence, the activity (phosphorylation level) of Akt and proteins that are known to associate with apoptosis was analyzed. After treating cells with various concentrations of arsenite for 24 h, the levels of phospho-Akt, Bcl2 and cytochrome c were analyzed. Fig. 2B shows that phospho-Akt and Bcl2 decreased while cytochrome c increased with the incremental arsenite concentrations. The effector caspase (caspase 3) activity was also analyzed. Fig. 2C shows that caspase 3 activity increases with the concentration of arsenite treatments, and the highest activity was achieved at 40 μM of arsenite. The addition of the apoptosis inhibitor (z-VAD-fmk) restored the dephosphorylated (active) form of Akt (Fig. 2D) and suppressed the caspase 3 activity in arsenite-treated cells (Fig. 2E). We also checked the fragmented DNA (sub-G1) fraction of the cells after arsenite treatments. The sub-G1 fraction increased in a dose-dependent manner with the treatments (Fig. S1†). These results indicate that arsenite treatment reduces Akt activity that culminates apoptotic death in HEK293 cells.
PTEN aggregates with arsenite treatment in HEK293 cells
PTEN (phosphatase and tensin homolog deleted on chromosome 10) is a negative regulator of the PI3K/Akt signal pathway. We therefore investigated whether the reduction of Akt activity is due to an increase in the PTEN level. Cells were treated with 0–60 μM of arsenite and the PTEN level was analyzed by immunoblotting. Noticeably, arsenite treatment reduced both PTEN (Fig. 3A) and phospho-Akt (Fig. 2B) levels. Apparently, arsenite acts independently on PTEN and Akt activities in HEK293 cells. In order to confirm whether PTEN is degraded with arsenite treatment, cells were boiled in SDS-containing buffer and the total cellular PTEN level was analyzed by immunoblotting. Noticeably, the total PTEN level was not reduced but slightly increased in cells treated with arsenite (Fig. 3B). Reportedly, arsenite stimulates protein aggregation in cells.29 We speculated that PTEN forms aggregates with arsenite administration so that the level reduced in the soluble fraction. The PTEN levels in the NP-40 soluble fraction (SF) and the insoluble fraction (InF) were then analyzed. Fig. 3A and C show that PTEN decreases in the SF but increases in the InF with the incremental arsenite concentrations. This finding indicates that PTEN aggregates with arsenite treatment. The accumulation of denatured PTEN in the cells implies a failure of the protein degradation system.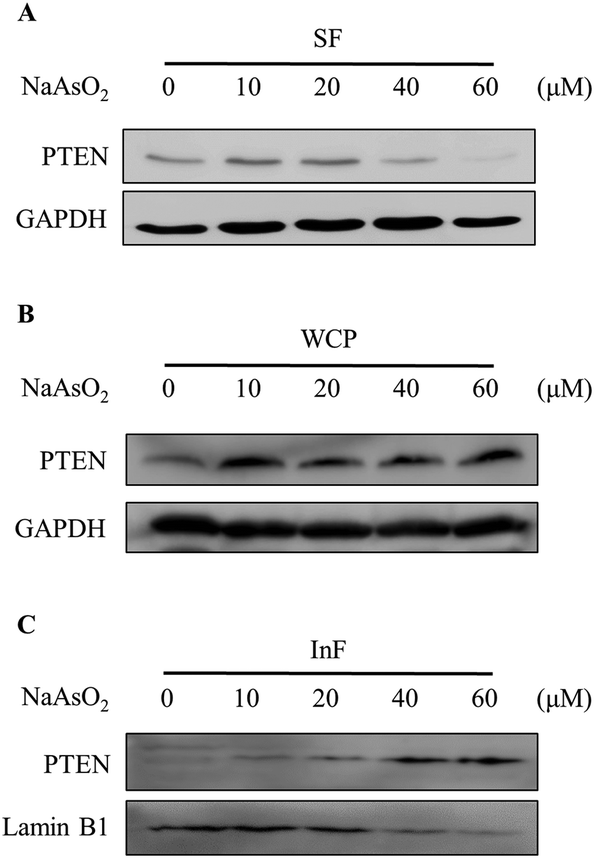 | ||
| Fig. 3 Cellular distribution of PTEN in arsenite-treated cells. HEK293 cells were treated with various concentrations of NaAsO2 for 24 h. PTEN levels in the soluble fraction (SF; A), the whole cell protein (WCP; B) and the insoluble fraction (InF; C) were analyzed by immunoblotting. | ||
Arsenite treatment activates autophagy-associated signaling factors but reduces lysosomal activity
The aggregated proteins in cells can be removed by an autophagic process through lysosomal degradation. It has been shown that arsenic treatment induces cell autophagy.24–26,28 Since PTEN accumulated in the InF with arsenite treatment, we examined the involvement of autophagy in arsenite-treated HEK293 cells. LC3 and p62 are markers for cells undergoing autophagy. The levels of LC3 and p62 were analyzed after arsenite treatment. LC3 (LC3-I and LC3-II) and p62 increased with arsenite administration in a dose- and time-dependent manner (Fig. 4B and C), indicating the occurrence of autophagy in the arsenite-treated renal cells.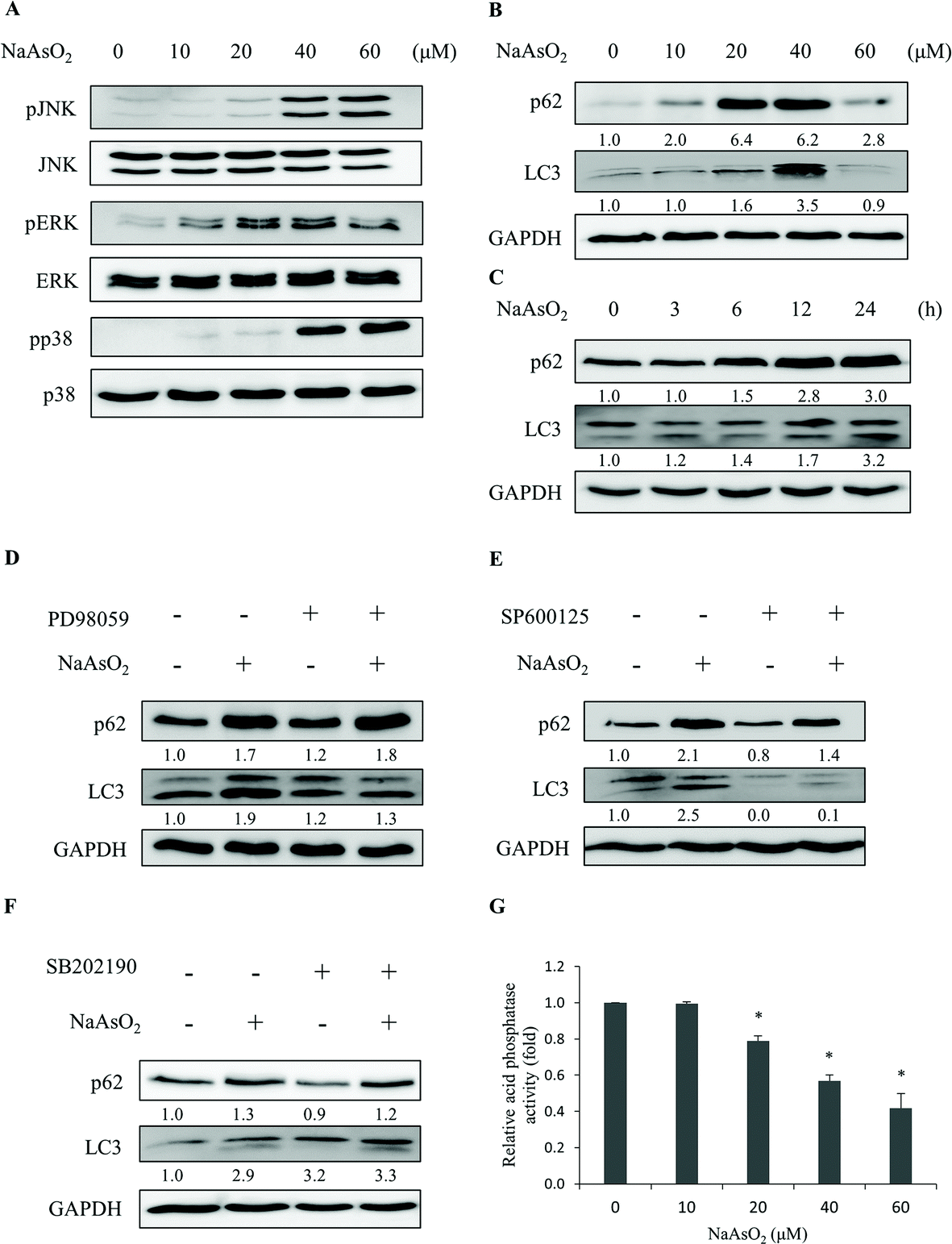 | ||
| Fig. 4 Arsenite treatments induce autophagy responses. HEK293 cells were treated with various concentrations of NaAsO2 for 24 h. Mitogen activated protein kinase activities with Akt, JNK, ERK, and p38 as target substrates (A) and markers (B) for autophagy (p62 and LC3) were analyzed. The p62 and LC3 levels were also determined for cells that were treated with 20 μM NaAsO2 for various time intervals (C). Cells were treated with 20 μM NaAsO2 for 24 h in the presence of the ERK (PD98059, 20 μM), JNK (SP600125, 15 μM) or p38 (SB202190, 10 μM) inhibitor and the p62 and LC3 levels were analyzed (D, E and F, respectively). The inhibitors were administered 30 min before adding the arsenite. Cells were treated with various concentrations of arsenite for 24 h and the lysosome (acid phosphatase) activity was determined (G). Each value represents a mean ± standard deviation of three independent samples. Asterisks indicate significant differences as compared to that of the untreated cells. *p < 0.05. GAPDH was used as a loading control to normalize the amount of sample applied for analysis (B–F). Numbers underneath the western blotting represent the amount of p62 or LC3 relative to that of the loading control. | ||
The occurrence of autophagy can be regulated via several signaling pathways.24–26 The potential signaling factors that can be activated by arsenite treatment were investigated. Cells were treated with 0–60 μM of sodium arsenite and the activities (the phosphorylated forms) of JNK, ERK, and p38 were examined. As shown in Fig. 4A, all these factors were activated with arsenite treatment. These results are contradictory to the decrease in Akt activity (Fig. 2B) in the same treatment. To further investigate the participation of various signaling factors in the arsenite-induced autophagy, the inhibitor of JNK (SP600125), ERK (PD98059) or p38 (SB202190) was added to the HEK293 cells. The results show that the addition of the ERK inhibitor reduced LC3 (Fig. 4D), while the addition of the JNK inhibitor reduced both LC3 and p62 levels (Fig. 4E) with arsenite treatment. On the other hand, the p38 inhibitor did not alter the LC3 or p62 level (Fig. 4F), suggesting the absence of the p38 signaling pathway in the arsenite-induced autophagy of HEK293 cells.
Despite the occurrence of autophagy with arsenite treatment, damaged proteins still accumulated in the cells. The autophagy is apparently blocked and thus cannot be completed. Possibly, lysosomal activity was inhibited by arsenite administration so that autophagy cannot be processed to completion. Lysosomal activity was thus examined. Acid phosphatase activity was analyzed to gauge the function of lysosomes. Fig. 4G shows that lysosomal activity reduced in a dose-dependent manner with arsenite treatment. The addition of ERK (PD98059), JNK (SP600125), p38 (SB202190) or apoptosis inhibitor (z-VAD-fmk) did not alter the arsenite-induced reduction of lysosomal activity (Fig. S2†). The data indicate that arsenite blocks the progression of autophagy by reducing the lysosomal activity.
Rapamycin stimulates the completion of autophagy via enhancing lysosomal activity in arsenite-treated cells
Lysosomal activity can be activated by mTOR inhibitors.30 The specific mTOR inhibitor, rapamycin, was added to the cells along with arsenite treatment. The analysis of the autophagy markers revealed a reduction in the p62 level with rapamycin administration (Fig. 5A). However, the LC3 level was not affected. Further analysis of the lysosomal activity indicated that rapamycin restored the arsenite-reduced lysosomal activity in a dose-dependent manner (Fig. 5B). The result demonstrates that rapamycin stimulates the lysosomal activity and accelerates the degradation of damaged proteins in arsenite-treated cells. Examination of the PTEN level in arsenite-treated cells showed a reduction of the protein in the InF with rapamycin treatment (Fig. 5C), implying an increase in lysosomal activity and autophagy.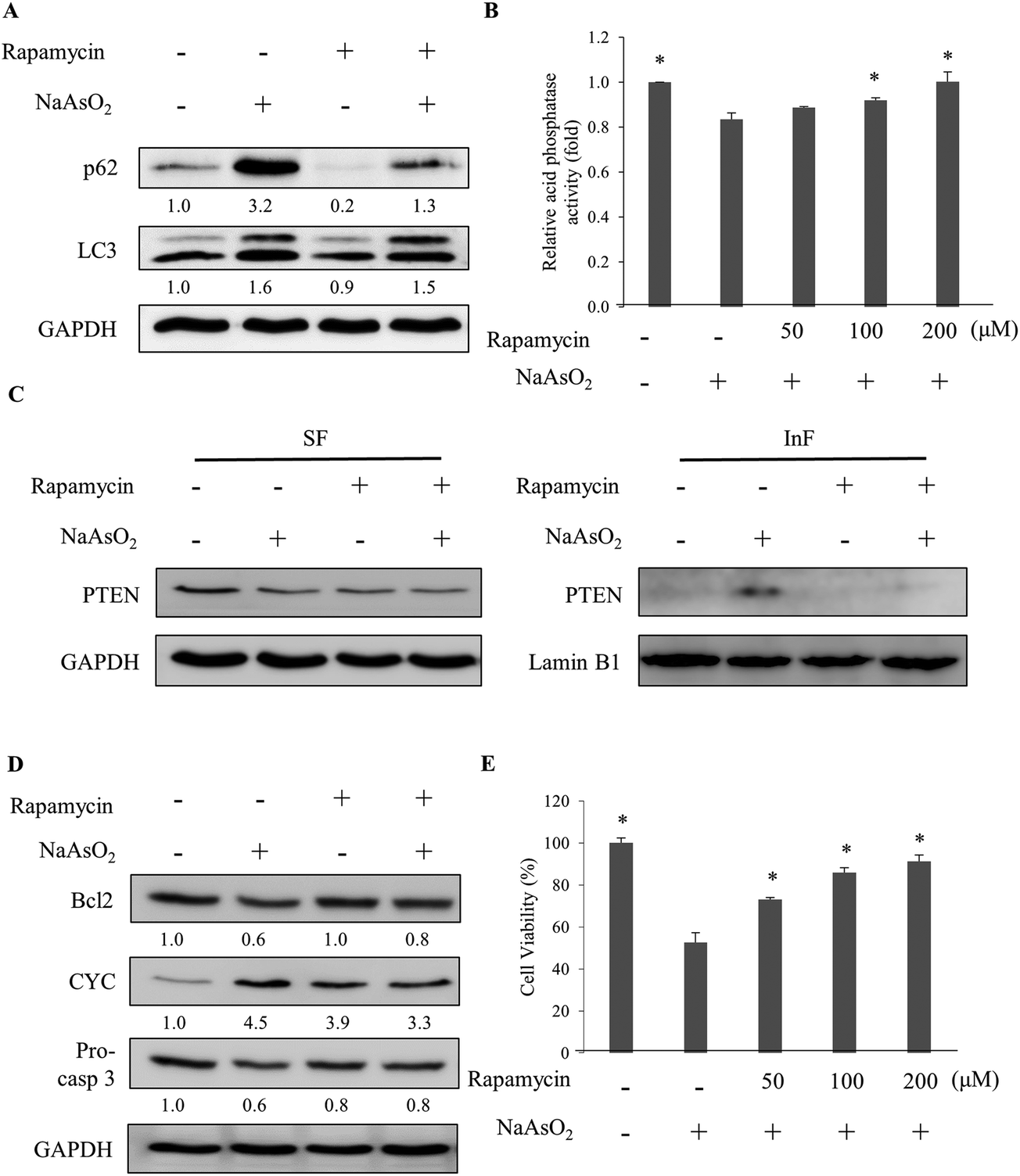 | ||
| Fig. 5 Rapamycin activates autophagy which is blocked by arsenite treatment. (A) HEK293 cells were treated with NaAsO2 in the presence of 100 nM rapamycin. The levels of p62 and LC3 were determined. (B) Cells were treated with NaAsO2 in the presence of various concentrations of rapamycin and the acid phosphatase activities were analyzed. (C) Cells were treated with arsenite in the presence of 100 nM rapamycin. The soluble (SF) and insoluble (InF) fractions of arsenite-treated cells were separated and the PTEN levels were analyzed by immunoblotting. (D) Cells were treated with NaAsO2 in the presence of 100 nM rapamycin. Bcl2, cytochrome c (CYC), and pro-caspase 3 (Pro-casp 3) levels were determined by immunoblotting. (E) Viability of the arsenite-treated cells in the presence of various concentrations of rapamycin. Rapamycin was administered to cells 30 min before the addition of 20 μM NaAsO2 and incubated for 24 h. Each value (B and E) represents a mean ± standard deviation of three independent samples. Asterisks indicate significant differences as compared to that of cells treated only with NaAsO2. *p < 0.05. GAPDH was used as a loading control to normalize the amount of the sample applied for analysis (A, C and D). Numbers underneath the western blotting represent the amount of p62 or LC3 relative to that of the loading control. | ||
Since rapamycin enhanced the activity of autophagy, the factors in the apoptosis pathway were further examined. Fig. 5D shows that rapamycin restored the Bcl2 and pro-caspase 3 levels but reduced the elevated level of cytochrome c. Since the inhibition of autophagy was attenuated by rapamycin, the stress generated by the accumulation of the damaged materials can be reduced. Cell viability is expected to increase with the rapamycin treatment. Examining the viability of arsenite-treated cells showed an increase of the survival fraction with rapamycin treatment in a dose-dependent manner (Fig. 5E). These results demonstrate that rapamycin reduced the arsenite-induced toxicity by increasing the lysosomal activity to allow the progression of autophagy.
Depletion of mTOR gene expression releases the blockade of autophagy and restores cell viability in arsenite-treated HEK293 cells
Recent studies show that mTOR inhibits the activity of autophagy while Akt modulates the mTOR activity.31,32 Since Akt activity reduced with arsenite treatment (Fig. 2B), we examined the mTOR activity (phospho-mTOR level) under arsenite exposure. Fig. 6A shows that mTOR activity reduced with the incremental arsenite concentration.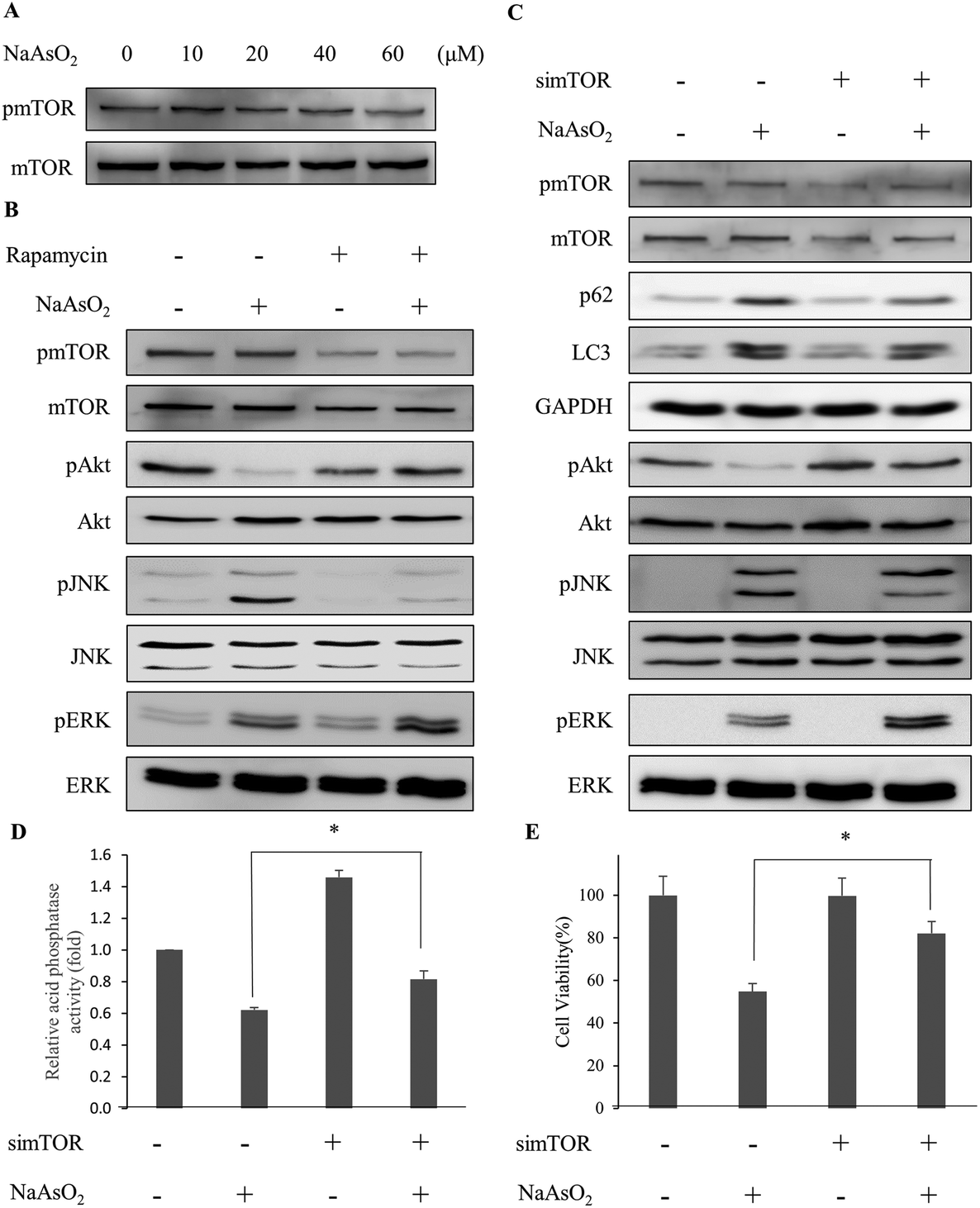 | ||
| Fig. 6 Depletion of mTOR activity rescues cells from arsenite-induced damage via the modulation of signaling factors involved in autophagy. (A) HEK293 cells were treated with various concentrations of NaAsO2 and the mTOR activity (represented by the level of phosphorylated mTOR and pmTOR) was analyzed by immunoblotting. (B) Cells were treated with 20 μM NaAsO2 for 24 h in the presence of 100 nM rapamycin. Signaling factors associated with autophagy were visualized with immunoblotting. Rapamycin was added 30 min prior to NaAsO2 treatment. (C) Expression of mTOR mRNA was knocked down by siRNA. Forty-eight hours after siRNA transfection, the cells were treated with 20 μM NaAsO2 for 24 h. Signaling factors associated with autophagy were visualized with immunoblotting. The mTOR knockdown cells were treated with 20 μM NaAsO2 for 24 h. Acid phosphatase activity (D) and cell viability (E) of the samples were determined. Each value represents a mean ± standard deviation of three independent samples. Asterisks indicate significant differences between the paired samples. *p < 0.05. | ||
The correlation of Akt with the autophagy-associated signaling factors was investigated. With the reduction of Akt activity (phospho-Akt level) by arsenite treatment, the addition of rapamycin restored the Akt activity in the arsenite-treated cells. In addition, ERK activity increased further but JNK activity decreased in arsenite-treated cells with rapamycin addition (Fig. 6B). Although the LC3 level was not significantly affected, the p62 level was reduced with rapamycin treatment (Fig. 5A).
To further clarify the role of mTOR, the mTOR mRNA expression was knocked down by siRNA. Fig. 6C shows that by knocking down the mTOR expression, Akt and ERK activities in arsenite-treated cells were increased. In contrast, JNK activity was decreased with the treatment. The observation is similar to that of rapamycin administration. The lysosomal (acidic phosphatase) activity (Fig. 6D) and cell viability (Fig. 6E) were increased after mTOR siRNA treatment. The data indicate that the arsenite-induced apoptotic effect was attenuated by knocking down the mTOR expression. These results indicate that the progression of autophagy is reactivated with the knockdown of mTOR expression and thus cytotoxicity is reduced resulting from the accumulation of damaged proteins in arsenite-treated cells.
Discussion
We established in this study the correlation of arsenite-induced autophagy and apoptosis in renal cells. Exposure to arsenite causes cellular protein denaturation. The denatured proteins cannot be effectively degraded and accumulate as insoluble aggregates. Akt, ERK and JNK signaling pathways are coordinately evoked to trigger the removal of damaged proteins via autophagy. Akt activity is reduced with arsenite treatment, resulting in a decrease in the mTOR activity. ERK is a downstream target of mTOR; its activity is negatively regulated by mTOR in autophagy. A decline in mTOR activity enhances the ERK activity that consequently increases the LC3 level. JNK is concurrently activated with arsenite treatment and positively regulates the p62 level. Both LC3 and p62 stimulate the formation of autophagosomes. The autophagosome is expected to fuse with the lysosome to form an autolysosome and the damaged materials can then be degraded by the lysosomal enzymes. However, arsenite treatment reduces lysosomal activity and inhibits the formation of autolysosomes. The blockade of autophagy triggers the activation of the apoptotic pathway and the cells then move toward apoptotic death. The depletion of mTOR activity enhances lysosomal activity in arsenite-treated cells and thus allows the progress of autophagy via the formation of autolysosomes. The recovery of autophagy facilitates the removal of damaged materials and reduces the stress caused by the accumulation of damaged materials. This release of strain restores Akt activity and attenuates the occurrence of apoptosis. This mechanism is illustrated in Fig. 7.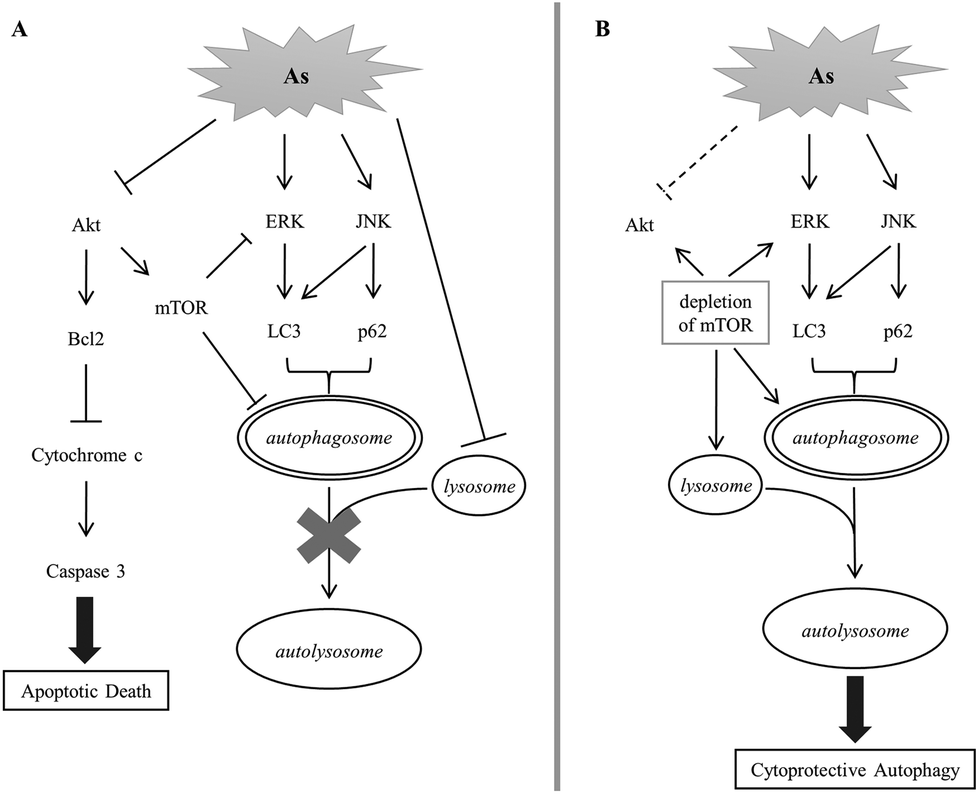 | ||
| Fig. 7 A postulated scheme for the signaling pathway of arsenite-induced autophagy and apoptosis in HEK293 cells. | ||
The accumulation of damaged proteins in cells can be cytotoxic. The degradation of denatured proteins or protein aggregates can be achieved through two pathways. One is via the ubiquitin–proteasome system. The aggregates are degraded by the 26S proteasome. The other is via the autophagosome–lysosome system (ALS) and the aggregates are degraded by the lysosome.33 Noticeably, the soluble PTEN level in arsenite-treated cells was not recovered in the presence of the proteasome inhibitor, MG132 (Fig. S3†). This result suggests that PTEN and other denatured proteins in arsenite-treated cells are degraded via the ALS.
Autophagy can be activated and employed to remove damaged materials that are produced by endogenous or exogenous stress. The proceeding of autophagy requires the binding of p62 to the damaged proteins and the recruitment of LC3 to the membrane of the autophagosome. The autophagosome then fuses with the lysosome to form the autolysosome.34,35 To complete this process, the inside of the lysosome has to maintain a pH lower than five with the help of the proton pumps located on the lysosomal membrane.36 The activity of the acid phosphatase is usually recognized as an indicator for lysosomal activity.37 Acid phosphatase is activated in an acidic environment and participates in the degradation of the damaged materials inside the autolysosome. With arsenite treatment, both LC3 and p62 increased in cells. Both proteins are degraded upon autolysosome formation.17,38 The persistent presence of both LC3 and p62 implies that the transition from the autophagosome to the autolysosome is inhibited. Additionally, acid phosphatase activity is reduced with arsenite treatment in a dose-dependent manner (Fig. 4G). These results indicate that the process of autophagy is blocked by arsenic administration. Owing to the blockade of autolysosome formation, the arsenite-induced protein aggregates accumulate in cells and activate the apoptosis signaling pathway.
We found in this study that arsenite-induced cell damage can be prevented by inhibiting the apoptotic pathway or by activating autophagy. The apoptosis inhibitor z-VAD-fmk reverts the pro-apoptotic and anti-apoptotic factors that are altered by arsenite treatment and restores the Akt activity. However, factors involved in autophagy (e.g., mTOR, LC3 and p62) are not altered by the pan-caspase inhibitor (Fig. S4†). Nonetheless, the depletion of mTOR elevates lysosomal activity and releases the blockade of autophagy in arsenite-treated renal cells. Consequently, Akt activity is restored and apoptotic activity is attenuated. Apparently, the autophagy process delivers a signal to activate Akt and inhibit apoptosis. Thus, Akt is a checkpoint to modulate the direction of the cells to autophagy or apoptosis. Factors that can restore Akt activity with arsenite exposure are potential detoxifiers of arsenic compounds. Noticeably, the apoptosis inhibitor (z-VAD-fmk) is not as potent as rapamycin to recover cell viability (Fig. 2A and 5E). Possibly, z-VAD-fmk blocks apoptosis but still cannot effectively remove the damaged materials. Cells may die through a non-apoptotic pathway.39
mTOR is also a critical factor in determining the fate of arsenite-exposed cells. For autophagy to occur, the mTOR activity has to be reduced. Akt is known to regulate the mTOR activity.31,32 Arsenic treatment causes a reduction in Akt and mTOR activities (Fig. 2B and 6A) and initiates autophagy. Reportedly, mTOR inhibitors (Torin1 and PP242) increase lysosomal activity.30 We show here that rapamycin has the same effect. Rapamycin can affect autophagy via dephosphorylation and activation of the ULK (Unc-51-like kinases) complex which stimulates the formation of autophagosomes. Rapamycin also stimulates the activity of the lysosome and hence enhances the efficiency of autophagy.40,41 Our results show for the first time that the depletion of mTOR activity attenuates arsenite-induced cytotoxicity.
Despite arsenite treatment reducing mTOR and lysosomal activities, further depletion of mTOR by rapamycin contrarily increases the lysosomal activity. Possibly, the activation of lysosomal activity is an additional effect of rapamycin besides mTOR inhibition. However, the knockdown of mTOR gene expression also enhanced the lysosomal activity in arsenite-treated cells (Fig. 6D). These results suggest that lysosomal activity is regulated by mTOR in the cells. We postulate that arsenite treatment reduces both mTOR and lysosomal activities. However, the degree of mTOR reduction is insufficient to stimulate the lysosomal activity. The mTOR activity has to drop below a certain threshold before lysosomes are activated and autophagy proceeded in arsenite-treated cells.
A variety of signaling pathways are activated by arsenite treatment. Arsenite induces autophagy via Akt inhibition and the MAPK signaling pathway activation.24–26 ERK, JNK and p38 are activated in different cell types treated with arsenic compounds. However, p38 does not participate in arsenite-induced autophagy in HEK293 cells (Fig. 4F) despite an increase in its activity (Fig. 4A). Noticeably, a mutual negative regulation exists between mTOR and ERK activities. Studies have indicated that the mTOR inhibitor activates ERK activity in several cell lines.42–44 Conversely, the activation of ERK reduced the mTOR activity in H4IIE (hepatoma), K562 (erythroleukemia) and MC3T3-E1 (osteoblast) cells with consequential autophagy.42,45 We demonstrated clearly in this study that mTOR is an upstream and negative regulator of ERK activity in arsenite-treated cells. Although we cannot delineate whether arsenite directly increases the ERK activity or achieves this goal via reducing the mTOR activity, rapamycin treatment or knockdown of mTOR expression synergistically increased ERK activity in arsenite-treated cells. Our results show clearly that mTOR inversely modulates the ERK activity which further regulates the level of LC3 and the formation of autophagosomes.
Besides ERK, JNK also participates in autophagy. JNK activity increases with anticancer drug (methotrexate and ciclopirox) treatment which induces the occurrence of autophagy; this effect can be blocked by the JNK inhibitor.46,47 With arsenite treatment, JNK is activated and the p62 level is increased (Fig. 4B and C). Administration of the JNK inhibitor reduces the p62 level. However, the inhibitor cannot alter the lysosomal activity (Fig. S2A†) or increase the cell viability (Fig. S5†). The data indicate that the reduction of JNK activity alone is not sufficient to relieve the blockade of autophagy in arsenite-treated cells. In addition, rapamycin or mTOR knockdown reduces JNK but enhances ERK activity (Fig. 6B and C). Co-administration of the ERK inhibitor with rapamycin did not rescue the viability of the as-treated cells (Fig. S6†). These results suggest that the enhancement of ERK and the reduction of JNK activities are indicators for the relief of the arsenite-induced blockade of autophagy.
In summary, we explore in this work the signaling pathway of arsenite-induced autophagy and apoptosis in renal cells. Arsenic treatment caused protein aggregation in renal cells and autophagy is activated to remove the damaged proteins. However, the autophagic process is blocked with the arsenite treatment which leads to the accumulation of the damaged materials in the cells. The failure to remove the denatured proteins keeps Akt in low activity which evokes the apoptotic cell death signaling pathway. The arsenite-damaged cells can be rescued by depleting the mTOR activity. Despite mTOR activity being reduced with arsenite treatment, further reduction of mTOR activity by rapamycin or mRNA knockdown is needed to activate the lysosomal activity and allow autophagy to proceed. The successful removal of the damaged materials by autophagy attenuates the reduction of Akt activity and increases the viability of the arsenite-treated cells.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grant MOST106-2514-S-007-002 from the Ministry of Science and Technology. The authors would like to thank Dr Ming F. Tam (Department of Biological Sciences, Carnegie Mellon University) for critical reading of the manuscript.References
- M. F. Naujokas, B. Anderson, H. Ahsan, H. V. Aposhian, J. H. Graziano, C. Thompson and W. A. Suk, The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem, Environ. Health Perspect., 2013, 121, 295–302 CrossRef CAS PubMed.
- I. W. G. o. t. E. o. C. R. t. Humans, Arsenic, metals, fibres, and dusts, IARC Monogr. Eval. Carcinog. Risks Hum., 2012, 100, 11–465 Search PubMed.
- F. Faita, L. Cori, F. Bianchi and M. G. Andreassi, Arsenic-induced genotoxicity and genetic susceptibility to arsenic-related pathologies, Int. J. Environ. Res. Public Health, 2013, 10, 1527–1546 CrossRef CAS PubMed.
- H. Wiseman and B. Halliwell, Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer, Biochem. J., 1996, 313(Pt 1), 17–29 CrossRef CAS PubMed.
- M. Bauer, M. Goldstein, M. Christmann, H. Becker, D. Heylmann and B. Kaina, Human monocytes are severely impaired in base and DNA double-strand break repair that renders them vulnerable to oxidative stress, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 21105–21110 CrossRef CAS PubMed.
- H. Shi, L. G. Hudson and K. J. Liu, Oxidative stress and apoptosis in metal ion-induced carcinogenesis, Free Radicals Biol. Med., 2004, 37, 582–593 CrossRef CAS PubMed.
- J. C. Reed, Apoptosis-targeted therapies for cancer, Cancer Cell, 2003, 3, 17–22 CrossRef CAS PubMed.
- A. Masood, A. S. Azmi and R. M. Mohammad, Small molecule inhibitors of bcl-2 family proteins for pancreatic cancer therapy, Cancers, 2011, 3, 1527–1549 CrossRef CAS PubMed.
- P. E. Czabotar, G. Lessene, A. Strasser and J. M. Adams, Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy, Nat. Rev. Mol. Cell Biol., 2014, 15, 49–63 CrossRef CAS PubMed.
- R. Altaf, M. Z. Asmawi, A. Dewa, A. Sadikun and M. I. Umar, Phytochemistry and medicinal properties of Phaleria macrocarpa (Scheff.) Boerl. extracts, Pharmacogn. Rev., 2013, 7, 73–80 CrossRef PubMed.
- E. White and R. S. DiPaola, The double-edged sword of autophagy modulation in cancer, Clin. Cancer Res., 2009, 15, 5308–5316 CrossRef PubMed.
- E. White, Deconvoluting the context-dependent role for autophagy in cancer, Nat. Rev. Cancer, 2012, 12, 401–410 CrossRef CAS PubMed.
- B. Levine and G. Kroemer, Autophagy in the pathogenesis of disease, Cell, 2008, 132, 27–42 CrossRef CAS PubMed.
- J. Debnath, E. H. Baehrecke and G. Kroemer, Does autophagy contribute to cell death?, Autophagy, 2005, 1, 66–74 CrossRef CAS PubMed.
- A. Kuma, M. Matsui and N. Mizushima, LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization, Autophagy, 2007, 3, 323–328 CrossRef CAS PubMed.
- S. Pankiv, T. H. Clausen, T. Lamark, A. Brech, J. A. Bruun, H. Outzen, A. Overvatn, G. Bjorkoy and T. Johansen, p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy, J. Biol. Chem., 2007, 282, 24131–24145 CrossRef CAS PubMed.
- I. Tanida, N. Minematsu-Ikeguchi, T. Ueno and E. Kominami, Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy, Autophagy, 2005, 1, 84–91 CrossRef CAS PubMed.
- A. Bhattacharjee, M. Hasanain, M. Kathuria, A. Singh, D. Datta, J. Sarkar and K. Mitra, Ormeloxifene-induced unfolded protein response contributes to autophagy-associated apoptosis via disruption of Akt/mTOR and activation of JNK, Sci. Rep., 2018, 8, 2303 CrossRef PubMed.
- V. M. Dan, B. Muralikrishnan, R. Sanawar, J. S. Vinodh, B. B. Burkul, K. P. Srinivas, A. Lekshmi, N. S. Pradeep, S. G. Dastager, B. Santhakumari, T. R. Santhoshkumar, R. A. Kumar and M. R. Pillai, Streptomyces sp metabolite(s) promotes Bax mediated intrinsic apoptosis and autophagy involving inhibition of mTOR pathway in cervical cancer cell lines, Sci. Rep., 2018, 8, 2810 CrossRef PubMed.
- K. Ding, H. Wang, Y. Wu, L. Zhang, J. Xu, T. Li, Y. Ding, L. Zhu and J. He, Rapamycin protects against apoptotic neuronal death and improves neurologic function after traumatic brain injury in mice via modulation of the mTOR-p53-Bax axis, J. Surg. Res., 2015, 194, 239–247 CrossRef CAS PubMed.
- Y. C. Kim and K. L. Guan, mTOR: a pharmacologic target for autophagy regulation, J. Clin. Invest., 2015, 125, 25–32 CrossRef PubMed.
- J. Jiang, J. Dai and H. Cui, Vitexin reverses the autophagy dysfunction to attenuate MCAO-induced cerebral ischemic stroke via mTOR/Ulk1 pathway, Biomed. Pharmacother., 2018, 99, 583–590 CrossRef CAS PubMed.
- E. A. Dunlop and A. R. Tee, mTOR and autophagy: a dynamic relationship governed by nutrients and energy, Semin. Cell Dev. Biol., 2014, 36, 121–129 CrossRef CAS PubMed.
- H. W. Chiu, S. Y. Ho, H. R. Guo and Y. J. Wang, Combination treatment with arsenic trioxide and irradiation enhances autophagic effects in U118-MG cells through increased mitotic arrest and regulation of PI3 K/Akt and ERK1/2 signaling pathways, Autophagy, 2009, 5, 472–483 CrossRef CAS PubMed.
- H. W. Chiu, Y. S. Ho and Y. J. Wang, Arsenic trioxide induces autophagy and apoptosis in human glioma cells in vitro and in vivo through downregulation of survivin, J. Mol. Med., 2011, 89, 927–941 CrossRef CAS PubMed.
- A. Nagappan, W. S. Lee, J. W. Yun, J. N. Lu, S. H. Chang, J. H. Jeong, G. S. Kim, J. M. Jung and S. C. Hong, Tetraarsenic hexoxide induces G2/M arrest, apoptosis, and autophagy via PI3 K/Akt suppression and p38 MAPK activation in SW620 human colon cancer cells, PLoS One, 2017, 12, e0174591 CrossRef PubMed.
- G. Wang, T. Zhang, W. Sun, H. Wang, F. Yin, Z. Wang, D. Zuo, M. Sun, Z. Zhou, B. Lin, J. Xu, Y. Hua, H. Li and Z. Cai, Arsenic sulfide induces apoptosis and autophagy through the activation of ROS/JNK and suppression of Akt/mTOR signaling pathways in osteosarcoma, Free Radicals Biol. Med., 2017, 106, 24–37 CrossRef CAS PubMed.
- H. W. Chiu, Y. C. Tseng, Y. H. Hsu, Y. F. Lin, N. P. Foo, H. R. Guo and Y. J. Wang, Arsenic trioxide induces programmed cell death through stimulation of ER stress and inhibition of the ubiquitin-proteasome system in human sarcoma cells, Cancer Lett., 2015, 356, 762–772 CrossRef CAS PubMed.
- T. Jacobson, C. Navarrete, S. K. Sharma, T. C. Sideri, S. Ibstedt, S. Priya, C. M. Grant, P. Christen, P. Goloubinoff and M. J. Tamas, Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast, J. Cell Sci., 2012, 125, 5073–5083 CrossRef CAS PubMed.
- J. Zhou, S. H. Tan, V. Nicolas, C. Bauvy, N. D. Yang, J. Zhang, Y. Xue, P. Codogno and H. M. Shen, Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion, Cell Res., 2013, 23, 508–523 CrossRef CAS PubMed.
- Q. M. Liang, Z. F. Luo, J. X. Zeng, W. Q. Chen, S. S. Foo, S. A. Lee, J. N. Ge, S. Wang, S. A. Goldman, B. V. Zlokovic, Z. Zhao and J. U. Jung, Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy, Cell Stem Cell, 2016, 19, 663–671 CrossRef CAS PubMed.
- J. P. Tsai, C. H. Lee, T. H. Ying, C. L. Lin, C. L. Lin, J. T. Hsueh and Y. H. Hsieh, Licochalcone A induces autophagy through PI3 K/Akt/mTOR inactivation and autophagy suppression enhances Licochalcone A-induced apoptosis of human cervical cancer cells, Oncotarget, 2015, 6, 28851–28866 Search PubMed.
- N. Chondrogianni, I. Petropoulos, S. Grimm, K. Georgila, B. Catalgol, B. Friguet, T. Grune and E. S. Gonos, Protein damage, repair and proteolysis, Mol. Aspects Med., 2014, 35, 1–71 CrossRef CAS PubMed.
- C. F. Bento, M. Renna, G. Ghislat, C. Puri, A. Ashkenazi, M. Vicinanza, F. M. Menzies and D. C. Rubinsztein, Mammalian Autophagy: How Does It Work?, Annu. Rev. Biochem., 2016, 85, 685–713 CrossRef CAS PubMed.
- S. Fulda and D. Kogel, Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy, Oncogene, 2015, 34, 5105–5113 CrossRef CAS PubMed.
- C. Settembre, A. Fraldi, D. L. Medina and A. Ballabio, Signals from the lysosome: a control centre for cellular clearance and energy metabolism, Nat. Rev. Mol. Cell Biol., 2013, 14, 283–296 CrossRef CAS PubMed.
- C. Y. Hsu and Y. L. Chuang, Changes in energy-regulated molecules in the trophocytes and fat cells of young and old worker honeybees (Apis mellifera), J. Gerontol., Ser. A, 2014, 69, 955–964 CrossRef CAS PubMed.
- P. D. Jiang and N. Mizushima, LC3-and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells, Methods, 2015, 75, 13–18 CrossRef CAS PubMed.
- W. J. Kim, R. R. Mohan, R. R. Mohan and S. E. Wilson, Caspase inhibitor z-VAD-FMK inhibits keratocyte apoptosis, but promotes keratocyte necrosis, after corneal epithelial scrape, Exp. Eye Res., 2000, 71, 225–232 CrossRef CAS PubMed.
- F. Demarchi, C. Bertoli, T. Copetti, I. Tanida, C. Brancolini, E. L. Eskelinen and C. Schneider, Calpain is required for macroautophagy in mammalian cells, J. Cell Biol., 2006, 175, 595–605 CrossRef CAS PubMed.
- J. Bove, M. Martinez-Vicente and M. Vila, Fighting neurodegeneration with rapamycin: mechanistic insights, Nat. Rev. Neurosci., 2011, 12, 437–452 CrossRef CAS PubMed.
- J. Wang, M. W. Whiteman, H. Lian, G. Wang, A. Singh, D. Huang and T. Denmark, A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1, J. Biol. Chem., 2009, 284, 21412–21424 CrossRef CAS PubMed.
- B. Hoang, A. Benavides, Y. Shi, Y. Yang, P. Frost, J. Gera and A. Lichtenstein, The PP242 mammalian target of rapamycin (mTOR) inhibitor activates extracellular signal-regulated kinase (ERK) in multiple myeloma cells via a target of rapamycin complex 1 (TORC1)/eukaryotic translation initiation factor 4E (eIF-4E)/RAF pathway and activation is a mechanism of resistance, J. Biol. Chem., 2012, 287, 21796–21805 CrossRef CAS PubMed.
- A. Carracedo, L. Ma, J. Teruya-Feldstein, F. Rojo, L. Salmena, A. Alimonti, A. Egia, A. T. Sasaki, G. Thomas, S. C. Kozma, A. Papa, C. Nardella, L. C. Cantley, J. Baselga and P. P. Pandolfi, Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer, J. Clin. Invest., 2008, 118, 3065–3074 CAS.
- Y. H. Yang, K. Chen, B. Li, J. W. Chen, X. F. Zheng, Y. R. Wang, S. D. Jiang and L. S. Jiang, Estradiol inhibits osteoblast apoptosis via promotion of autophagy through the ER-ERK-mTOR pathway, Apoptosis, 2013, 18, 1363–1375 CrossRef CAS PubMed.
- Y. Shen, J. Yang, J. Zhao, C. Xiao, C. Xu and Y. Xiang, The switch from ER stress-induced apoptosis to autophagy via ROS-mediated JNK/p62 signals: A survival mechanism in methotrexate-resistant choriocarcinoma cells, Exp. Cell Res., 2015, 334, 207–218 CrossRef CAS PubMed.
- H. Zhou, T. Shen, C. Shang, Y. Luo, L. Liu, J. Yan, Y. Li and S. Huang, Ciclopirox induces autophagy through reactive oxygen species-mediated activation of JNK signaling pathway, Oncotarget, 2014, 5, 10140–10150 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tx00238j |
| This journal is © The Royal Society of Chemistry 2019 |