
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe association between chemical-induced porphyria and hepatic cancer
Andrew G.
Smith
 *a and
John R.
Foster
b
*a and
John R.
Foster
b
aMRC Toxicology Unit, Hodgkin Building, University of Leicester, Lancaster Road, Leicester LE2 4UA, UK. E-mail: ags56@mrc-tox.cam.ac.uk
bToxPath Sciences Ltd, 1 Troutbeck Avenue, Congleton, Cheshire, CW12 4JA, UK
First published on 1st June 2018
Abstract
The haem biosynthetic pathway is of fundamental importance for cellular metabolism both for the erythroid and nonerythroid tissues. There are several genetic variants of the pathway in the human population that cause dysfunction of one or other of the enzymes resulting in porphyrias of varying severity. Serious chronic hepatic and systemic diseases may result. Some of these can be precipitated by exposure to drugs including hormones, barbiturates and antibiotics, as well as alcohol and particular chlorinated aromatic chemicals. In experimental animals some of the steps of this pathway can also be severely disrupted by a variety of environmental chemicals, potential drugs and pesticides, especially in the liver, leading to the accumulation of uroporphyrins derived from the intermediate uroporphyrinogens or protoporphyrin IX, the immediate precursor of haem. With some of these chemicals this also leads to cholestasis and liver cell injury and eventually hepatic tumours. The review evaluates the available evidence linking hepatic porphyria with carcinogenesis in naturally occurring human genetic conditions and in chemically-induced porphyrias in laboratory animals. The existing data showing gender, strain, and species differences in sensitivity to the chemical-induced porphyrias, liver injury and liver tumours are discussed and the role that transgenically altered mouse models have played in defining the varying mechanisms. Finally, the review proposes a novel, unifying hypothesis linking the hepatotoxicity induced by the accumulation of various porphyrins, with the increased risk of developing hepatic cancer as a long term consequence.
1. Introduction
The hepatic toxicities of a variety of drugs, pesticides and environmental chlorinated chemicals in animals include marked disturbances of the haem biosynthetic pathway.1–4 The formation of haem in mammals is an essential pathway accounting for much of the use of iron either for transport and storage of oxygen, or for its use in cytochromes of respiration, oxygenases and signalling. Most of the haem synthesized in the body is used in the bone marrow for haemoglobin and about 10% for intracellular metabolism. However, although virtually all cells require haem, the liver accounts for the greatest amount of non-erythrocyte haem formation, not only for mitochondrial function and steroid, bile acid and prostaglandin synthesis, but particularly in the metabolism of drugs, xenobiotics and plant constituents. The hepatic pathway is very responsive to the demand for haem as in circadian rhythm and in induction of cytochrome P450, whilst turnover can be quickly stimulated by the action of inducible haem oxygenase 1 (HMOX1). In humans, dominant or recessive genetic variants of the pathway can lead to porphyrias, malfunctions and accumulation of intermediate precursors of haem some of which may be toxic (including oxidized product porphyrins), and have serious clinical outcomes.5 The genetic penetrance can be very low and the clinical disorder may be precipitated by exogenous factors such as alcohol or by changes in physiological demand for haem controlled by hormones or nutrition.4 Some chemicals cause porphyria in rodents similar to types of human porphyrias without the requirement of predisposing gene variants of haem synthesis.3,4The incidences of liver cancer are higher than expected in surveys of patients with some types of clinical porphyria where the numbers are sufficient for valid studies. Many of the chemicals that cause porphyria in rodents cause eventually hepatocellular tumours. This review focuses on some drugs, herbicides and chlorinated aromatic chemicals that cause both porphyria and liver tumours or precancerous changes in rodents and compares with types of human porphyrias in which associations with increased incidences of liver cancer have been described.
2. Interference of haem synthesis by chemicals
The biosynthesis of haem involves eight steps in which simple precursors from the tricarboxylic acid (TCA) cycle are built into the complex macrocyclic molecule, protoporphyrin IX, into which iron is then incorporated (Fig. 1). Both in the liver and red blood cells, the first step is the condensation between glycine and succinyl CoA to give 5-aminolaevulinate (5-ALA). Condensation differs between non-erythroid and erythroid tissue being catalyzed by different 5-aminolaevulinate synthetase enzymes (ALAS1 and ALAS2) regulated by available haem or iron pools respectively.6 Particular enzymes of the haem pathway in the liver are affected by a variety of chemicals both directly and indirectly (Fig. 1 and Table 1). Some are susceptible to direct inhibition by metal ions, drugs, herbicides or their metabolites. Other enzymes are inhibited by xenobiotic metabolites generated endogenously from haem precursors after stimulation of xenobiotic responsive pathways, some of which still need a more complete understanding. Inhibition of a particular enzyme can lead to accumulation of intermediate porphyrinogens, and their oxidized product porphyrins, and can be compounded by feedback stimulation of the rate controlling enzyme ALAS1. When inhibition of the haem pathway occurs, it is the pathological accumulation of hepatic porphyrins in rodent liver, by some of these drugs and other chemicals, that is causal in inducing hepatic toxicity. The toxicity may not be due entirely to the porphyrins per se; porphyria and liver injury may also be the outcome of common mechanisms, probably of an oxidative nature. While carcinogenicity studies have been carried out for some of these porphyrogenic chemicals, many may not have been tested in cancer studies. Most of the porphyrogenic chemicals that have been shown to cause liver tumours in rodents are associated with hepatic toxicity and are thought to be carcinogenic by non-genotoxic mechanisms, perhaps as a consequence of their cellular toxicity, of which the key initiating events (KIE) may have been hypothesized but not yet elucidated.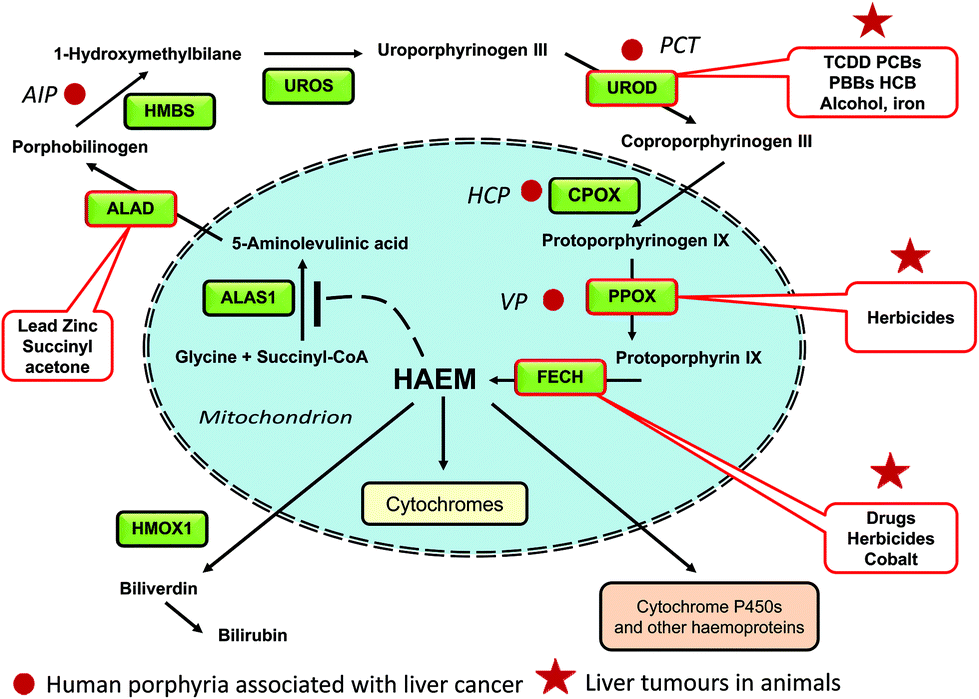 | ||
| Fig. 1 Nonerythrocyte haem synthesis showing enzymic steps that are affected by chemicals and drugs in vivo leading to malfunctions of haem production. ALAS1, 5-aminolevulinic acid synthase; ALAD, aminolevulinic acid dehydratase; HMBS, 1-hydroxymethybilane synthase; UROS, uroporphyrinogen III synthase; UROD, uroporphyrinogen III decarboxylase; CPOX, coproporphyrinogen III oxidase; PPOX protoporphyrinogen IX oxidase; FECH, ferrochelatase; HMOX1, haem oxygenase 1. AIP, acute intermittent porphyria; HCP, hereditary coproporphyria; PCT, porphyria cutanea tarda; VP, variegate porphyria. | ||
| Chemical | Main porphyrin involveda | Nuclear receptor? | Molecular MOA | Hepatotoxicity | Species | Carcinogenic response (incidence) | Ref. |
|---|---|---|---|---|---|---|---|
| a Does not include elevation of plasma and hepatic 5-ALA which may be substantial in some experimental and human acute porphyrias. N/A Not applicable. | |||||||
| Rodent chemical porphyrogen | |||||||
| 3-[2-(2,4,6-Trimethylphenyl)-thioethyl]-4-methylsydnone (TTMS) | Protoporphyrin IX | CAR/PXR | Ferrochelatase inhibition | Wt↑; inflammation; necrosis; hepatocellular hypertrophy | Mouse | Unknown | 87 and 88 |
| Tralkoxydim | Protoporphyrin IX | CAR/PXR | Ferrochelatase inhibition | Wt↑; inflammation; necrosis; hepatocellular hypertrophy; bile duct hyperplasia; porphyrin | Mouse ≫ rat = hamster ≫ human | (−ve rat, hamster) Not conducted in mouse! | 87, 94 and 100 |
| Griseofulvin | Protoporphyrin IX | CAR/PXR | Ferrochelatase inhibition | Wt↑; inflammation; necrosis; hepatocellular hypertrophy; porphyrin | Mouse M > F ⋙ human | ∼90% mouse (−ve in rat, hamster) | 85, 86, 98, 104 and 105 |
| 3,5-Diethoxycarbonyl-1,4-dihydro-2,4,6-trimethylpyridine (DDC) | Protoporphyrin IX | CAR/PXR | Ferrochelatase inhibition | Wt↑; inflammation; necrosis; hepatocellular hypertrophy in mouse, rat and chick, porphyrin | Rat = mouse = chick | Unknown | 2, 80 and 81 |
| Fomesafen | Protoporphyrin IX | N/A | Inhibition of PPOX | ALT↑ liver wt↑ in mouse; porphyrin | Mouse ICR | Yes; mouse (16% HCC; 100% AHF) | 77 |
| Human & rodent chemical porphyrogen | |||||||
| Polychlorinated biphenyls (PCBs) | Uroporphyrin | AHR agonist | Inhibition of UROD | Wt↑; inflammation; necrosis, hypertrophy; porphyrin | Rat = mouse ≫ human | Yes; mouse > rat | 36 and 56 |
| 2,3,7,8-Tetrachorodibenzo-p-dioxin (TCDD) | Uroporphyrin | AHR agonist | Inhibition of UROD | Wt↑; inflammation; necrosis, hypertrophy; porphyrin | Rat = mouse ≫ human | Yes; mouse > rat | 3, 34, 35, 54 and 121 |
| Hexachlorobenzene (HCB) | Uroporphyrin | AHR agonist? | Inhibition of UROD | Wt↑; inflammation, necrosis; hypertrophy; porphyrin | Rat = mouse ≫ Human | Yes; mouse > rat | 37–39, 54 and 122 |
| Human genetic disorders | |||||||
| Acute intermittent porphyria (AIP) | Porphobilinogen | N/A | Hydroxymethylbilane synthase (HMBS) deficiency | Drug-induced severe hepatitis | Human | Yes | 9 and 14–21 |
| Porphyria cutanea tarda (PCT) | Uroporphyrin | N/A | Uroporphyrinogen decarboxylase (UROD) deficiency | Mild-moderate hepatitis | Human | Yes (5–12% of patients) | 10 and 25–31 |
| Variegate porphyria (VP) | Porphobilinogen | N/A | Protoporphyrinogen oxidase (PPOX) deficiency | Moderate hepatitis | Human | Yes | 21 |
| Erythropoietic porphyria (EPP) | Protoporphyrin IX | N/A | Ferrochelatase (FECH); less commonly 5-aminolevulinic acid synthase-2 (ALAS2) | Drug-induced hepatitis | Human | Yes | 5, 78 and 117 |
| Hereditary coproporphyria (HCP) | Porphobilinogen; coproporphyrin | N/A | Deficiency in coproporphyrinogen oxidase (CPOX) | Drug-induced hepatitis | Human | Yes | 5, 11 and 13 |
3. Hepatocellular cancer in porphyria patients
All but one of the enzyme steps of non-erythrocyte haem biosynthesis are associated in humans with rare, but clinically established, phenotypes of porphyria or related disorders which are caused by recessive variants, or autosomal dominant variants, triggered by endogenous or exogenous factors.4,5 A disorder associated with genetic variants of ALAS1 is not known, possibly because either it would be lethal, or because of the potential for the gene to be upregulated to compensate for any deficiency in enzyme activity (Fig. 1). For some types of porphyria, particularly acute intermittent porphyria (AIP), and porphyria cutanea tarda (PCT), higher incidences of primary liver cancer in patients than a reference population have long been reported from different countries.7–10In patients with acute hepatic porphyrias, AIP, variegate porphyria (VP) and hereditary coproporphyria (HCP) (due to variants in the HMBS, PPOX, and CPOX, genes respectively, see Fig. 1), acute neurovisceral attacks are triggered by the accumulation of porphyrin precursors in the circulation. Many individuals carrying these mutations will remain asymptomatic but can be triggered into disease after physiological stimuli such as changes in hormones or stress, or following the administration of porphyrogenic drugs such as sex hormones, barbiturates, and sulfonamide antibiotics. The acute attacks are probably associated with the neurotoxicity of highly elevated levels of 5-ALA produced in the liver.5,11–13 However porphobilinogen, uroporphyrin III, coproporphyrin III and protoporphyrin IX, derived from the malfunctioning haem pathway, also accumulate in the liver depending on the type of acute porphyria. In VP and HCP these haem precursors may contribute to the liver injury, but they also provoke dermal photosensitivity due to deposition in the skin.
Over the last few decades it has become clear that the acute hepatic porphyrias are associated with a markedly increased risk of patients developing primary liver cancer (HCC), at least for Northern European countries and without underlying cirrhosis. This was first described for AIP from northern Sweden9 and now confirmed in a number of other independent studies of not only AIP but also VP and HCP in countries such as Sweden, Norway, France and Switzerland. Investigations on Finnish and Swedish cohorts with AIP reported increased risk ratios for HCC compared to reference populations of up to about 100 times. It is unclear why reports are only from European countries but this may simply reflect health data organizational arrangements and clinical interest.14–20 Study of an AIP and of a VP patient has suggested that as well as the underlying HMBS and PPOX germline mutations, the hepatic tumour tissue also had second-hit somatic mutations in the respective genes.21 Whether these second mutations occurred incidentally or were instrumental in the subsequent development of the hepatic cancer is unknown and their role in the chronic elevation of heme precursors such as 5-ALA, porphobilinogen and porphyrins, remains unclear. It is possible that 5-ALA is pro-oxidant and genotoxic in hepatocytes22 but perhaps other mechanisms may operate such as dysfunction of the mitochondria if haem synthesis is severely disturbed. What does seem clear is that, like clinical AIP, the development of HCC is more likely to occur in women than in men. In addition, unlike most cases of HCC, the incidence of cirrhosis is low and without other major findings of underlying liver disease in these porphyric patients.23
Type 1 tyrosinemia is a genetic disease of tyrosine metabolism in which fumarylacetoacetate hydrolase (FAH) is inefficient and some of the neurological and other symptoms resemble those of AIP. Side reactions of accumulating fumarylacetoacetate and maleylacetoacetate form succinylacetone, a potent inhibitor of aminolevulinate dehydratase (ALAD) (Fig. 1). Patients can be treated with nitrisinone (2-(2-nitro-trifluoro-methylbenzoyl-1,3-cyclohexadione) originally developed as a pesticide) restricting precursors for FAH.136 If untreated there is a high incidence of liver damage and HCC in these patients.137
PCT patients show marked inhibition of hepatic uroporphyrinogen decarboxylase activity (UROD) with mild or moderate liver damage and an increased susceptibility to developing liver cancer (Fig. 1). Under uv light the uroporphyrins and other porphyrins arising from disruption of the haem pathway fluoresce red in the liver due to their deposition. Porphyrins released into circulation are deposited elsewhere in the body and can cause marked cutaneous photosensitivity. Unlike the acute porphyrias there is no marked rise in plasma or urinary 5-ALA and no neurotoxicity. Autosomal dominant inheritance with low penetrance of a UROD gene variant is a contributing factor for familial form of PCT. However, most patients have a sporadic form with no apparent association with adverse variants of the gene.5 Consumption of alcohol, oestrogenic drugs, other chemicals and hepatitis viruses have all been reported as precipitating agents for PCT, even for the familial form which may occur at a younger age.3 Most of the PCT patients are diagnosed at middle age or older. This is the most common type of clinical porphyria in many populations. Iron metabolism is implicated in the pathogenesis of PCT and depletion of body iron stores is one treatment approach. For some patient cohorts inheritance of the haemochromatosis HFE variant has been a strong risk factor in the development of the porphyria.24 The role of iron is not really understood, other than possibly promoting reactive oxygen species, but must clearly be the consequence of genetic variants in iron mobilization perhaps releasing Fe2+. This has been discussed in more detail elsewhere.3 A disorder very similar to PCT occurred in many people, particularly children, poisoned by hexachlorobenzene (HCB) and there is evidence that other highly chlorinated aromatic chemicals such as the 2,3,7,8-tetrachorodibenzo-p-dioxin (TCDD) and polychlorinated biphenyls (PCBs) have a similar potential.3
As with the acute porphyrias, there is clear evidence of an association between PCT and the risk of developing HCC.10,25–30 In one of these studies, 342 patients with PCT were observed over a 15-year period, 5% of them died from primary liver cancer.31 In another study, 6 out of 78 patients with HCC had PCT. Most studies have confirmed the early findings while others have been less positive, probably because of the complex nature of PCT development, and the possible confounding role of concurrent cirrhosis. Recognition of PCT and subsequent treatment may be resulting in a decrease in the likelihood of patients progressing to HCC. However, the subject is of interest in consideration of the risk assessment of cancer associated with exposure to environmental levels of polychlorinated aromatic chemicals.
4. Hepatic tumours in rodents caused by porphyria-inducing chemicals
Although metals such as lead and mercury are known to interfere in haem synthesis, both in liver and in erythrocytes, the resulting clinical chemical signs from human and experimental studies are usually porphyrinurias, frequently increased coproporphyrin I or III, or excretion in stool of protoporphyrin rather than overt porphyrias.1 Sometimes these reflect relatively mild disturbances of haem synthesis. What appears to be especially important in determining the onset, or otherwise, of hepatic porphyrias is the particular enzyme in the pathway that is affected and permits the accumulation of toxic haem precursors. In lead poisoning there is inhibition of haem biosynthesis but the metal has been shown to inhibit ALA dehydratase resulting in increased urinary 5-ALA without affecting porphobilinogen or resulting in the hepatic build-up of toxic porphyrins. Hence while lead is known to cause porphyrinuria with an increased excretion of coproporphyrins, its inability to inhibit markedly later enzymes prevents the onset of hepatic porphyrias and their dire consequences.135 Drugs and chemicals that trigger AIP in humans do not do so in animals without genetic models particularly the disrupted HMBDS gene.32,33Some of the chemicals and drugs known to cause disturbance of hepatic haem synthesis in animals, and sometimes in humans, induce not only overt porphyrias in rodents but also induce hepatocellular cancer, or forms of chronic liver injury which might predispose to cancer.
4a. Chemicals causing hepatic uroporphyria
HCB, dioxins and PCBs have been known for many years to cause hepatic symptoms in rodents similar to human PCT with inhibition of UROD (probably through an oxidative sequence involving oxidation of uroporphyrinogen),34,35 porphyria and liver injury.3 As with human patients there is strong evidence for a synergistic role of iron metabolism, and genetic predispositions. One key mechanistic aspect to these chemicals for porphyria seems to be through their binding to, and activation of, the aryl hydrocarbon receptor (AHR) with associated induction of hepatic CYP1A enzymes.3 For chemicals, such as chlorinated dioxins and dioxin-like PCB constituents of commercial PCB mixtures such as Aroclor 1254 and 1260, porphyria potency can be ascribed partly to the toxicity equivalence compared to the prototype AHR ligand, TCDD.36 However, for HCB the evidence is less clear.37 Hahn et al. concluded that porphyria induced by HCB is caused by weak AHR binding activity of the chemical38,39 and a genetic locus for susceptibility in mice corresponds to variants of the Ahr gene.40 World Health Organization views did not regard HCB as an AHR ligand and noted that the chemical could be contaminated by dioxins.36 This has been a four decade debate. In fact, the particular commercial HCB source used by many research laboratories for porphyria studies was chosen originally for its known high purity as a chemical standard41,42 and shown to have CYP1A inducing properties in rodents.43,44 Up to now the levels of contaminants present in this source have not been fully reported. The levels of TCDD equivalents due to PCDDs and PCDFs present in this HCB are now shown in Table 2. Comparison with previous porphyria experiments suggest the levels are too low to account significantly for in vivo hepatic porphyrogenic activity of HCB in rodents and that probably it can act as a weak AHR ligand.| PCDDs or PCDFs in HCB | Concentration | WHO TEF (2005) | TEQ concentration |
|---|---|---|---|
| ng g−1 of solid | ng g−1 of solid | ||
| a Organic Standard grade HCB for elemental analyses was from B.D.H. Ltd, UK. and analysed for A.G.S. (2005) at the Central Science Laboratory, DEFRA, York, UK by high resolution gas chromatography with high resolution mass spectrometry for PCDDs and PCDFs using 13C-labelled analogues for standardisation. Results were UKAS accredited. WHO Toxic Equivalent Factors (TEFs)36 were used to calculate Toxic equivalents to TCDD (TEQs) with upper values of ranges. Comparison of TEQs present in HCB with doses of TCDD that cause porphyria in rats and mice showed levels were insufficient to account for the responses.46,59,133,134 | |||
| 2,3,7,8-TCDD | 88.00 | 1 | 88.0 |
| 1,2,3,7,8-PeCDD | <0.59 | 1 | 0.6 |
| 1,2,3,4,7,8-HxCDD | <0.59 | 0.1 | 0.6 |
| 1,2,3,6,7,8-HxCDD | <0.94 | 0.1 | 0.1 |
| 1,2,3,7,8,9-HxCDD | <0.71 | 0.1 | 0.1 |
| 1,2,3,4,6,7,8-HpCDD | 18.94 | 0.01 | 0.2 |
| OCDD | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 011 011 |
0.0003 | 4.8 |
| 2,3,7,8-TCDF | <1.03 | 0.1 | 0.1 |
| 1,2,3,7,8-PeCDF | <0.47 | 0.03 | 0.01 |
| 2,3,4,7,8-PeCDF | <4.35 | 0.3 | 0.1 |
| 1,2,3,4,7,8-HxCDF | 2.82 | 0.1 | 0.3 |
| 1,2,3,6,7,8-HxCDF | 1.76 | 0.1 | 0.2 |
| 1,2,3,7,8,9-HxCDF | <1.06 | 0.1 | 0.1 |
| 2,3,4,6,7,8-HxCDF | <1.41 | 0.1 | 0.1 |
| 1,2,3,4,6,7,8-HpCDF | 1060 | 0.01 | 10.6 |
| 1,2,3,4,7,8,9-HpCDF | 30.6 | 0.01 | 0.3 |
| OCDF | 5978 | 0.0003 | 1.8 |
| Total | 108 | ||
In subchronic studies in rats HCB, TCDD, PCB and PBB mixtures have all shown a marked preponderance of females compared to males in inhibition of hepatic UROD activity and developing porphyria.45,46 Interestingly, this marked sexual dimorphism in porphyria also occurs in chronic studies of the development of liver tumours with these chemicals (Fig. 2).47–53 This is in contrast to a range of other genotoxic and nongenotoxic hepatocarcinogens. With HCB, the hepatotoxicity gives rise to peliosis and necrosis with haemosiderosis which may increase tumourgenic potential.54 The development of hepatocellular cancer in rats has been of some interest for the risk assessment of polyhalogenated aromatic chemicals such as TCDD and PCBs. The promotion of rodent liver hepatic carcinogenesis by these chemicals has been investigated and discussed in detail55–57 but the reason for initiation of tumourgenesis has not been explored fully although a number of earlier hypotheses sought to explain the predominance of tumours in females. Initiation with diethylnitrosamine (DEN) followed by TCDD or other polyhalogenated chemicals has often been used to model the complete neoplastic process.57 Comparison of hepatic tumours in the rat, caused by HCB, with hepatic tumours from rats first given an initiating dose of DEN followed by a promoting regimen of HCB, showed markedly different results. There was no consistent sex difference in timing or severity with the initiation-promotion regimen.58 When tissue from the two regimens was compared under uv light for uroporphyrin fluorescence as a consequence of hepatic porphyria the distribution of porphyrins between tumour and non tumour areas were very different (Fig. 3).
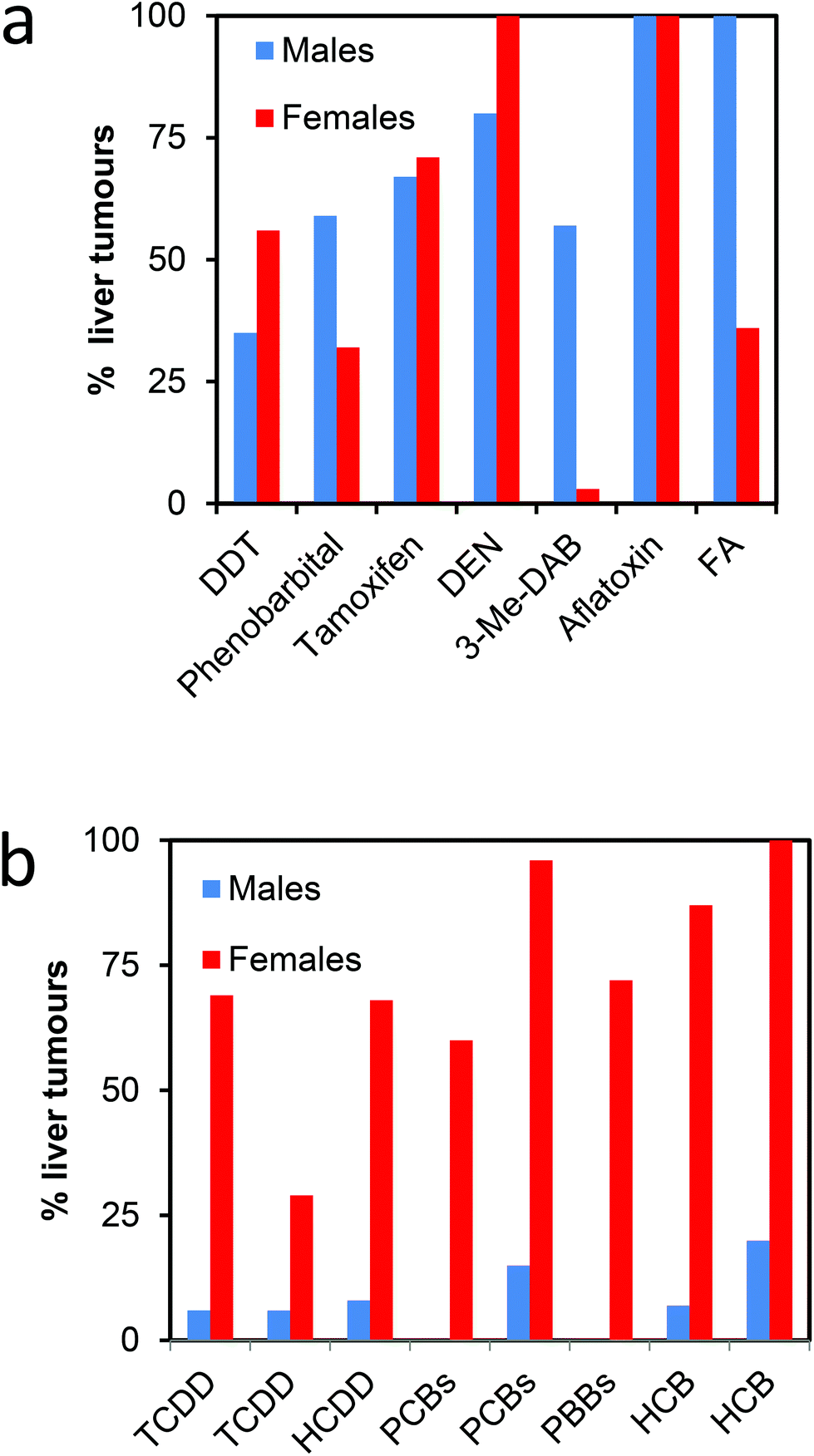 | ||
| Fig. 2 Incidences of liver tumours in toxicological studies of male and female rats. (a). Selection of some genotoxic and nongenotoxic carcinogens.126–131 (b). Polyhalogenated aromatic chemicals that also cause hepatic porphyria.47–53 Additional references of hepatocarcinogens can be found in Smith (2003).139 DEN, diethylnitrosamine, FA or AAF, N-2-fluorenylacetamide or 2 acetylaminofluorene; 3-Me-DAB, 3′-methyl-4-dimethylaminoazobenzene, tamoxifen and aflatoxin may be regarded as genotoxic carcinogens; DDT and phenobarbital may be regarded as nongenotoxic. | ||
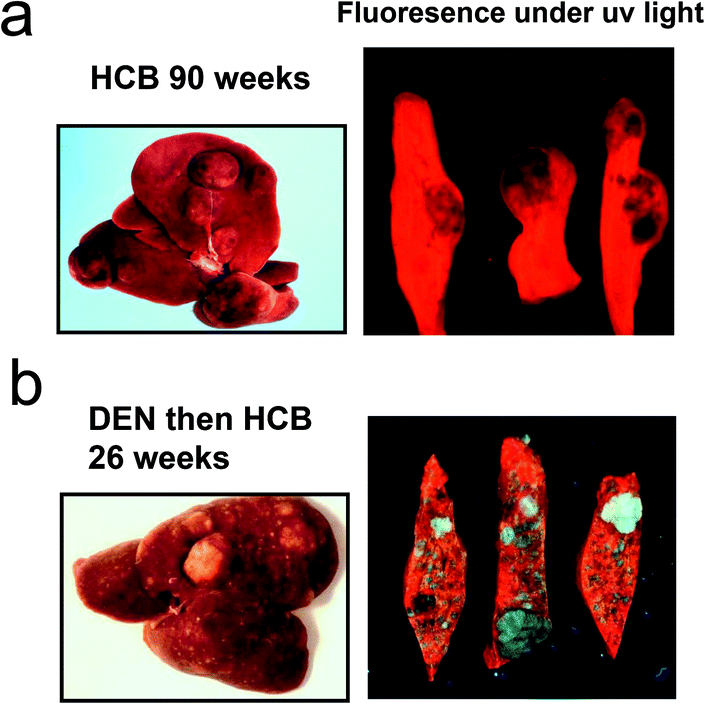 | ||
| Fig. 3 Difference between liver tumours in female rats caused by HCB alone and in those initiated with diethylnitrosamine (DEN) and promoted with HCB. (a) Female rats were fed HCB in diet (0.02%) for 90 weeks.47 (b) Female rats received DEN in the drinking water (0.015%) for 3 weeks then after 2 weeks’ rest were administered HCB in the diet (0.02%) for 30 weeks.58 Sections of liver were examined under uv light to demonstrate red fluorescence of uroporphyrin. Studies were conducted originally under the authorizations of UK Home Office regulations in the MRC toxicology unit. | ||
Given the evidence for a role of iron metabolism in PCT it was of interest that HCB porphyria in female rats seemed to be associated with iron status,47,59 and also CYP1A2 inducibility.46 Iron overload moderately enhanced hepatic carcinogenesis induced by HCB alone.60 In other studies with aflatoxin B1 and DEN, iron overload had no, or a reducing, effect on the incidence of liver carcinogenesis in rats (A. G. Smith, unpublished data). Thus rat studies might suggest a link between porphyria, perhaps inducing cytotoxicity, and liver cancer caused by polyhalogenated aromatic chemicals, and which may have a parallel in human PCT.
In mice, the development of porphyria by TCDD, PCBs and HCB was found to be influenced profoundly by hepatic iron status during experiments designed to explore mechanistic models of human PCT.3 Administration of iron not only enhanced porphyria, and hepatic toxicity, in mice with an AHR showing high affinity for TCDD, but overcoming resistance in some mouse strains with low affinity AHR.61,62 Subsequent studies demonstrated that there are genetic loci in mice independent of the AHR gene that contribute to the development of porphyria and hepatic toxicity.63 The genes responsible have not yet been identified but we would suggest some of them are associated with iron mobilization and use. Importantly, in chronic experiments with the AHR responsive strain C57BL/10ScSn, not only were the porphyria and hepatic toxicity inducing properties of HCB and PCBs markedly potentiated by prior iron treatment but so too was the subsequent incidence of liver adenomas and cancer to a marked degree.64,65 In contrast, the low affinity AHR DBA/2 strain also possessing other resistant loci was highly refractory to both porphyria and hepatocarcinogenesis following iron and PCBs. CYP1A2 gene null C57BL/6J mice with no CYP1A2 expression (mostly under control of the AHR) are highly resistant to porphyria caused by TCDD and HCB, even after iron loading pretreatment.66,67 The exact role of CYP1A2 in the porphyria mechanisms is still unclear although postulated to be in uroporphyrinogen oxidation. CYP1A2 knockout mice did not develop hepatocellular foci or adenomas following treatment with PCBs68 Octachlorostyrene, which only causes mild porphyria in iron-loaded C57BL10ScSn mice, was only weakly tumourgenic in contrast to HCB in the same mouse strain.69 Other nongenotoxic carcinogens in mice such as phenobarbital and nafenopin, were unaffected by iron pretreatment. Hepatocellular tumors resected from mice following treatment with HCB and PCBs did not show increased incidences of Ha-ras mutations but did show the emergence of a mononucleated diploid population consistent with precancerous changes.70,71 Tumour explants derived from the induced hepatocellular cancers could be passaged subcutaneously many times in syngeneic mice and showed many similarities whether of HCB or PCB origins. Homozygous knockout of the P53 gene in mice had little influence on the liver tumour incidence. Uroporphyria in rodents and PCT are likely due to an oxidative mechanism. Increased oxidative DNA damage was detected in C57BL/10ScSn mice after treatment with PCBs.3,72 Point mutations could not be detected using an in vivo mutation system suggesting that more complex chromosomal deletions or rearrangement are involved in the oxidative damage or in the mutagenicity of the porphyrins.73 However, other mechanisms associated with disturbed production of haem could be involved but are yet to be understood.
4b. Protoporphyrinogen oxidase inhibitors
Some chemicals that are inhibitors of protoporphyrinogen IX oxidase (PPOX) in green plants have been formulated as herbicides for post emergence weeds.74 Early products were diphenyl ethers, although later generation herbicides are often based on non-oxygen bridge compounds. The protoporphyrinogen that accumulates in an unregulated way is light sensitive and is oxidized chemically, rather than enzymically, to protoporphyrin IX which may catalyse oxygen free radical-induced damage in the photosynthetic plant cell. These herbicides can have similar inhibitory properties in mammals and although the protoporphyrin IX formed in the liver is not exposed to light, it still has hepatotoxic potential. The toxicity of these PPOX inhibitors in animals might be considered as having some similarities with variegate porphyria (Fig. 1) but the risk to humans from herbicide use has been considered to be very low due to their low exposure, rapid metabolism and excretion. However, chronic administration to rodents of a diphenyl ether PPOX inhibitor, fomesafen, has been studied particularly with respect to its possible mammalian porphyria inducing properties and toxicity.75,76 Interestingly, in iron treated C57BL/6 mice fomesafen enhanced preneoplastic changes in the liver such as frequency of altered foci and nodules.774c. Drugs and pesticides that inhibit ferrochelatase
In humans with erythropoietic porphyria (EPP) the enzyme ferrochelatase that catalyzes the incorporation of iron into protoporphyrin IX is compromised and some patients have severe hepatic disease probably reflecting mostly the deposition of toxic erythrocytic protoporphyrin from the blood into the liver (Table 1).5,78 There are several chemicals that have been shown to induce a similar porphyria in rodents and other animals through inhibition of hepatic ferrochelatase (Fig. 1). These chemicals are of a variety of types but are common in their ability to convert the haem in some liver cytochrome P450s (CYPs) into N-alkylated porphyrins which can be strong inhibitors of ferrochelatase.2,79 As well as porphyria these drugs and pesticides often cause cholestasis and hepatic toxicity in animals, which may lead to neoplastic lesions, but this can be species specific and has been the subject of risk assessment concerns.The generalized molecular mechanism of induced protoporphyria with these types of chemicals involves suicidal inactivation of cytochrome P450 with loss of associated enzyme activities (Fig. 4).2,80,81,94 Reactive metabolites from the parent molecules bind covalently to the prosthetic haem of the cytochrome through N-alkylation of one of the pyrrole moieties. The alkylated haem then dissociates from the apoprotein and the iron atom is liberated to yield N-alkylprotoporphyrin IX compounds (Fig. 5). The subsequent loss of haem results in a feedback upregulation of hepatic ALAS1.82,83 Stimulation of this enzyme drives increased haem synthesis compounding the inhibition of ferrochelatase and adding to the subsequent hepatic accumulation of porphyrin. In porphyria induced by 3,5-diethoxycarbonyl-1,4-dihydro-2,4,6-trimethylpyridine (DDC), cytochrome P450 mediated metabolism generates a metabolite from which a 4-methyl substituent is transferred onto the pyrrole nitrogen atom of the haem molecule and the alkylated product, N-methyl protoporphyrin, is a powerful inhibitor of ferrochelatase.79,84 In mice fed (1% of diet) the antifungal agent griseofulvin, hepatic ferrochelatase activity was decreased to 24% after 3 days. Concomitantly, the ALAS1 activity was increased by 6.6 fold.85,86
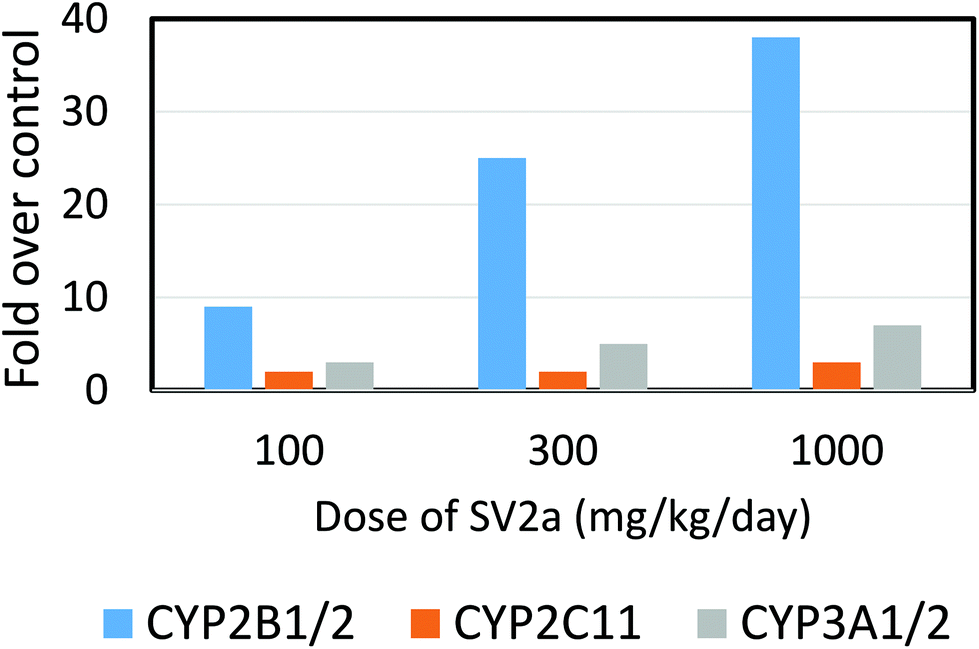 | ||
| Fig. 4 Effect of a porphyrinogenic agent, an anti-epileptic synaptic vesicle 2a (SV2a) ligand, 2-[4-(2-chloro-2,2-difluoroethyl)-2-oxopyrrolidin-1-yl]butanamide, in causing depressed activity of hepatic phenobarbital-type CYP2B family enzyme in the dog. Data are means from 4 male and 4 female dogs following 4 weeks’ administration. Redrawn from Nicolas et al. (2014).94 CYP2B11 was induced by the prodrug but at the highest dose given reduced enzyme activity was observed (but not the apoprotein by immunoblot). At the highest dose ferrochelatase was inhibited, hepatic protoporphyrin IX levels were highly elevated, N-alkylated porphyrins were detected, and hepatic transaminases in the plasma were markedly elevated indicating liver injury. Rats dosed with even higher amounts of the ligand (up to 1000 mg kg−1 day−1) showed much higher induction of CYP2B enzyme activities but no decrease at the highest dose and only 2-fold increase in porphyrins and very small elevations of plasma transaminases. | ||
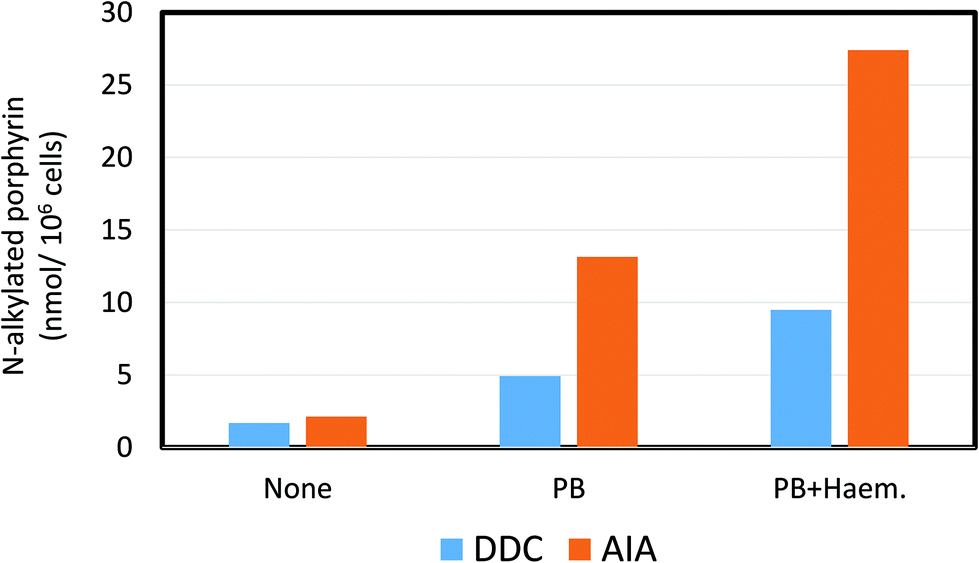 | ||
| Fig. 5 Effect of the ferrochelatase inhibiting porphyrinogenic agents, 3,5-diethoxycarbonyl-1,4-dihydro-2,4,6-trimethylpyridine (DDC) and allylisopropyl acetamide (AIA) on the N-alkylated porphyrin content of isolated rat hepatocytes. PB = the effect of pre-dosing in vivo with phenobarbital on the production of N-alkylated porphyrins by increasing cytochrome P4502B content. PB + Haem = the effect of pre-exposure with phenobarbital and haematin (to provide additional source of haem) on the production of N-alkylated porphyrins. Drawn from data in De Matteis et al. (1982).132 | ||
Other chemicals inducing protoporphyria or causing N-alkylated porphyrins in rodents include the anti-arthritic drug, 3-[2-(2,4,6-trimethylphenyl)-thioethyl]-4-methylsydnone (TTMS), ethylene, diethylnitrosamine, norethindrone, and the herbicides, tralkoxydim, ATMP, and ETC.87–92
The chemical-induced protoporphyria is associated with significant liver weight increases in susceptible animal species and although the actual cause of the weight increases has not been definitively ascribed, it is most probably multifactorial involving a combination of microsomal CYP induction, hepatic inflammation and necrosis, and hepatocellular hypertrophy.93,94 Dependent upon the degree of hepatopathy (liver pathology), the weight increases are frequently accompanied by increases in plasma alanine aminotransferase (ALT), alkaline phosphatase, aspartate aminotransferase and, mostly dependent upon the animal species involved, γ-glutamyl transferase.87,94 Although none of these enzyme biomarkers are diagnostic of the porphyria they reflect the degree of liver damage that may be associated with accumulation of protoporphyrin. In some studies, elevations in bilirubin can also be seen indicating a degree of cholestasis associated with the porphyrin-induced liver damage.94,95 In mice fed griseofulvin characteristic gene expression profiles were associated with disruption of lipid metabolism, and the pathologic states of liver toxicity, inflammation, early fibrosis, and cholestasis.86
Histopathologically the initial accumulation of the protoporphyrin IX occurs as red “Maltese cross” accumulations within the hepatocytes and Kupffer cells. Prolonged dosing of the porphyrogenic chemicals leads to progressive accumulation within the bile canaliculi between hepatocytes, the appearance of crystalline forms of the protoporphyrin IX within distended and hypertrophic bile ducts (Fig. 6a),87,94,96,97 and hepatocyte degeneration and inflammation, all of which are most severe in the periportal regions of the liver lobules (Fig. 6c).87,98 The protoporphyrin IX deposits appear birefringent under polarized light and display a red fluorescence under ultraviolet light (Fig. 6b and d). Of particular importance, with regard to the subsequent development of neoplasia by some of these porphyrogenic chemicals, is that induction of mitotic activity in the hepatocytes occurs between 2 to 5 days following administration of griseofulvin in the food97 and, while not recorded with other porphyrogenic drugs, would be expected to have occurred from the increased liver weight that almost invariably occurs following exposure to high doses of these porphyrogenic chemicals. The increase of mitotic activity in the mouse liver correlates with the increase in liver weight over time reaching 4.2-fold concurrent control liver weight after 4 to 6 months of feeding of griseofulvin at a concentration of 2.5% in the diet. Although few in number, where histological investigations are available of humans with porphyria, in PCT the pattern of pathology is similar to that described in the livers of mice given griseofulvin with the prominent presence of needle-like porphyrin crystals (although uroporphyrin not protoporphyrin), hepatocellular necrosis and regenerative hyperplasia.25
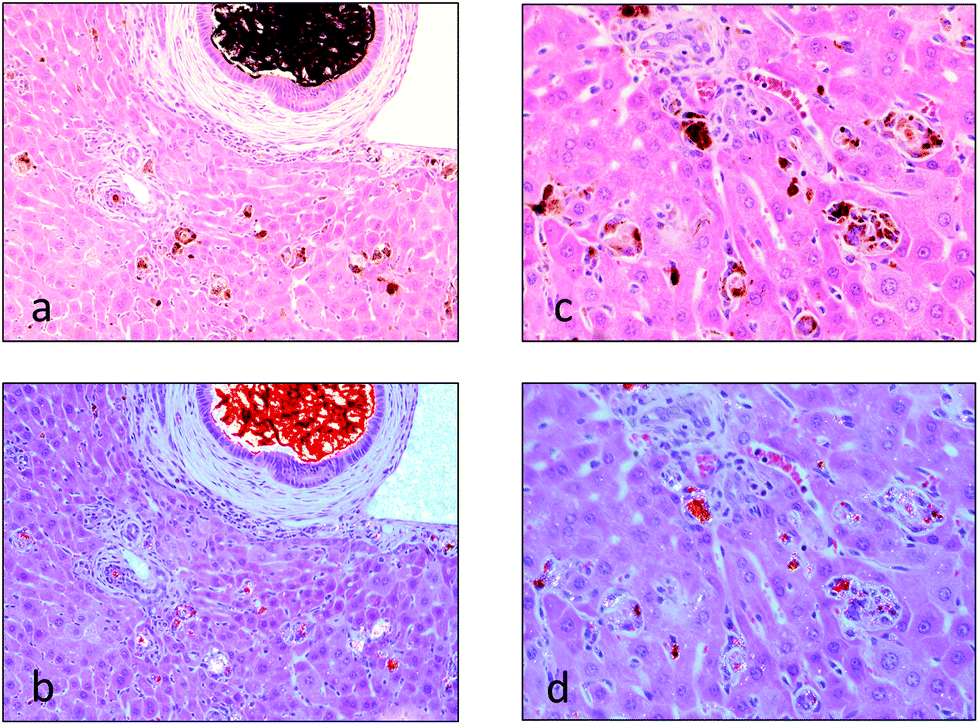 | ||
| Fig. 6 Periportal area in liver from a mouse given tralkoxydim for 28 days showing distended bile duct with hypertrophic lining mucosa (×20 microscope magnification) (a and b) and bile duct and phagocytic cells with brown pigment accumulation in hepatocytes and phagocytic cells (×40 magnification) (c and d). Viewed by conventional light (a and c) and polarized light demonstrating birefringent red porphyrin crystals (b and d). Liver sections were from studies conducted under the authorizations of UK Home Office regulations. | ||
While some of these chemicals appear to induce porphyria irrespective of animal species, others show distinct strain, species and sex differences in sensitivity, with griseofulvin being more toxic to male than female mouse liver, and tralkoxidim being a potent porphyrogen in the mouse but not in the rat or hamster.83,95,99,100 Similarly an antiepileptic, synaptic vesicle 2a (SV2a) ligand, drug candidate was found to form a CYP mediated N-alkylprotoporphyrin derivative that inhibited ferrochelatase in the dog but not in the rat nor in human hepatocytes in vitro (Fig. 4).94 The species difference operating in this instance is the metabolic ability to generate the reactive metabolite needed to alkylate protoporphyrin IX in the dog as opposed to the insensitive species. In contrast, the porphyrinogenic drug, DDC, induces porphyria as a result of ferrochelatase inhibition in rats, mice and chick livers,2 while TTMS induces porphyria in rats, mice and dogs.87 For those chemicals that induce protoporphyria in one or more animal species the outcome appears to be largely, if not solely, determined by the rate of generation of the appropriate CYP dependent hepatic metabolite.101 Those species that fail to generate appropriate amounts of the alkylated protoporphyrin will not inhibit ferrochelatase leading to protoporphyria. This has been adequately demonstrated for tralkoxydim which induces protoporphria in the mouse in vivo, and in mouse hepatocytes in vitro, but not in rat in vivo nor in vitro, nor human hepatocytes.102 Clearly these species differences are important in consideration of extrapolation to human risks.
In a survey of drug-induced liver injury in humans in Taiwan,103 of a total of 90847 patients taking one or more fungicides, including griseofulvin, eight were reported to have suffered hepatic injury, necessitating termination, following treatment with high doses of griseofulvin. Increased porphyrin has been reported in humans given high doses of griseofulvin but it is normally mild in comparison with that seen in the mouse model.93 In rare cases where the drug was taken at high doses over prolonged periods of time liver damage and serious skin reactions were reported accompanying the protoporphyria.93 Evidence supporting the drug-induced production of porphyria in man includes a survey of patients receiving griseofulvin and compared to a further group who had been off treatment for between 2–80 weeks.104 Hence griseofulvin is not recommended for patients suffering pre-existing porphyrias.105
Porphyrins are reported to be generators of reactive oxygen species, they affect hepatic redox balance and lead to induction of the antioxidant defense enzyme systems such that mice treated with griseofulvin show increased activity of glutathione reductase, superoxide dismutase, glutathione-S-transferase, and increased levels of reduced glutathione and malondialdehyde. In contrast the hepatic activities of glutathione peroxidase and catalase were reduced following griseofulvin administration.106 Griseofulvin has also been shown to reduce bile flow as a result of direct liver damage and effects on the biliary system, leading to the hepatocytes accumulating toxic endogenous bile acids such as taurocholic and taurodeoxycholic acid.107
Hepatocellular carcinomas, at incidences up to 90%, have been observed in mice fed griseofulvin at maximum concentrations of 3% in the diet for periods of up to 120 weeks.95,108,109 As well as causing accumulation of porphyrin within the liver, griseofulvin also induces necrosis and inflammation in a dose dependent fashion.106,110 Lifetime studies with griseofulvin, of similar duration to those in mice, in the rat and hamster have not shown the development of liver cancer and neither do these two species show the protoporphyria following shorter duration exposure.93 It is of note that the NOEL (approximately 0.1% in the diet) for griseofulvin induced liver enlargement and porphyria in mice111,112 is similar to the NOEL for tumor induction in mice (0.3% in the diet).109 To complicate factors further, hepatocellular carcinomas have been reported to develop in male mice offspring suggesting that irreversible nuclear changes had occurred after as little as 21 days of exposure.
Lifetime carcinogenicity studies on tralkoxydim have not been carried out in mice, which show protoporphyria after subchronic studies. It is not known if mice would have developed liver cancer after chronic exposure. Carcinogenicity studies have been conducted in rats and hamsters even though porphyria is not seen in sub-chronic studies in either the rat or the hamster. In neither species were hepatocellular neoplasms observed following lifetime exposure to tralkoxydim. The prediction from these studies is that tralkoxydim would be a liver carcinogen in mice if lifetime studies had been carried out, and that the absence of hepatocarcinogenicity in the non-porphyrinogenic rat and hamster is a consequence of the lack of porphyria induced by tralkoxydim in these species.100
5. Evidence for pathogenesis from mutated mice
Direct evidence that disruption of haem biosynthesis and the resulting porphyrin-induced hepatopathy can cause hepatic tumours in rodents, has come from genetically modified mice. BALB/c Fech(m1/Pas1) mice that have a radiation-induced missense mutation in the ferrochelatase gene (resulting in ∼6% activity of wild type), have markedly increased hepatic and serum protoporphyrin IX levels. They also have severe liver toxicity as evidenced by elevated plasma transaminases and bilirubin, cholestasis and pathological evidence of hepatic degeneration and inflammation (Fig. 7).113,114 In less than a year, these mice develop hepatocellular tumors in the absence of chemical treatment, demonstrating that porphyria alone can be carcinogenic to the liver.114,115 These mice also show depressed expression of total cytochrome P450 and many of its isoforms in comparison to their expression in wild-type mice. A related mouse model, more pertinent of human EPP, and carrying a targeted mutated ferrochelatase gene, showed significantly enlarged livers and spleens, and a high grade hepatobiliary pathology with a prominent accumulation of protoporphyrin IX in the biliary canaliculi and portal macrophages116 very similar to that shown in human porphyric patients with a pronounced liver pathology.117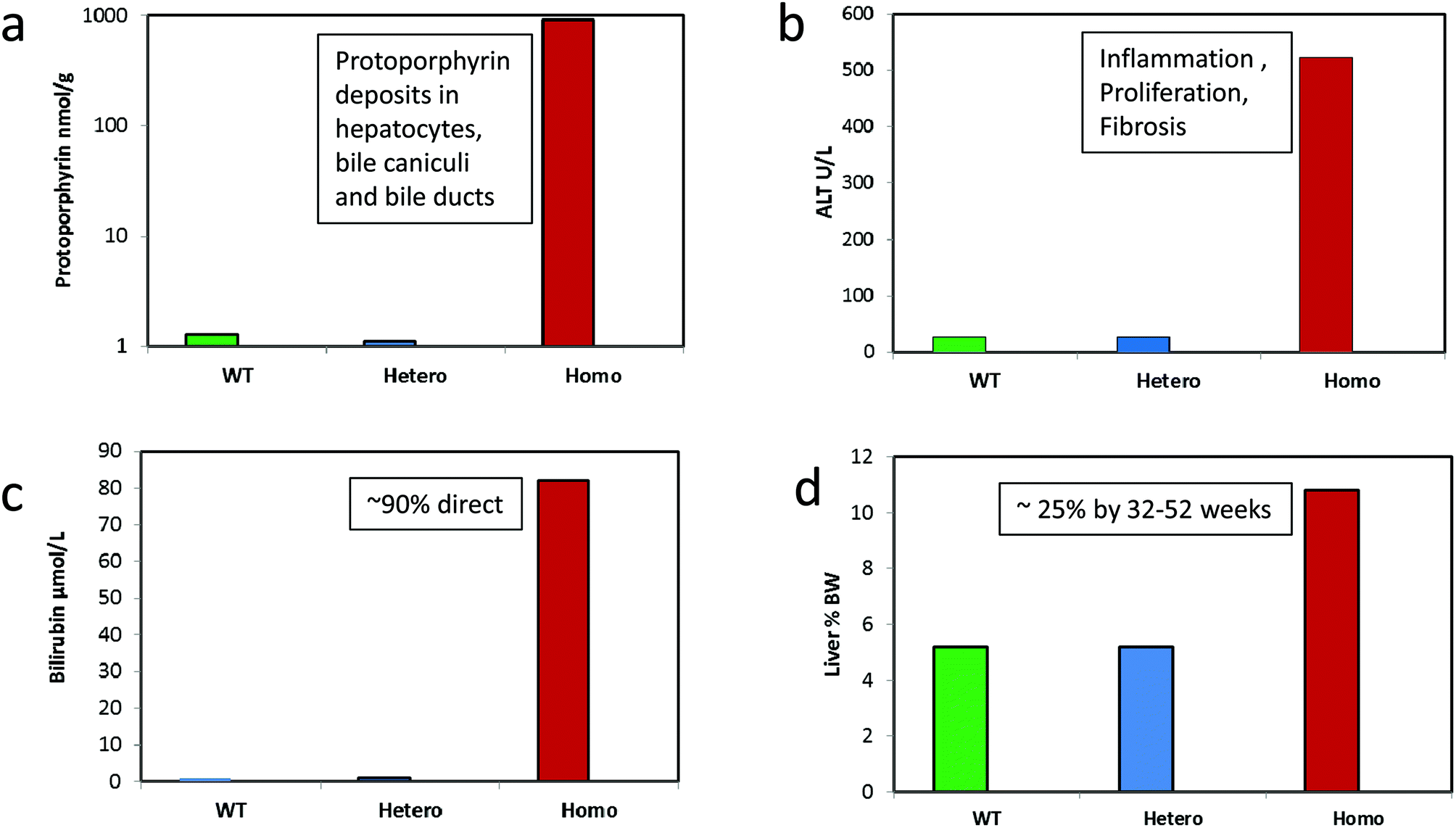 | ||
| Fig. 7 Effect of severely depressed hepatic ferrochelatase activity causing protoporphyria and hepatic toxicity in homozygote BALB/c Fech(m1/Pas1) mice carrying a mutated ferrochelatase gene. (a) Hepatic protoporphyrin levels, (b) plasma transaminase (ALT) levels, (c) plasma total bilirubin levels, (d) liver weight relative to body weight. Data are 8 weeks old male mice from Davies et al. (2005) descended from those described by Tutois et al. (1991) and produced as an F2 cross from wild type BALB/C and mutant mice (6 WT, 14 heterozygotes, 5 homozygotes).113,114 Studies were conducted originally in the MRC toxicology Unit under the authorization of UK Home office regulations. Homozygous mice eventually developed eosinophilic and clear cell foci, adenomas and adenocarcinomas. | ||
Mitoferrin 1 protein is a mitochondrial iron transporter critical for haem synthesis. In a mouse model with a deleted mitoferrin 1 gene, animals fed 5-ALA to stimulate haem synthesis showed increased liver weight in comparison to wild type mice, with homozygotes having appreciably higher liver weights than the heterozygotes.118 Livers from heterozygous mice showed bile duct proliferation and chronic inflammation in the periportal area with a marked accumulation of protoporphyrin IX pigment in the bile ducts of the 5-ALA-administered mutated mice that was not present in wildtype mice treated similarly. The heterozygous mice had a cholestatic pathology and early fibrosis which was more severe in the homozygous mice. Fibrosis was bridged between the portal and centrilobular areas with the development of hyperplastic liver nodules. Hence in this model, in the presence of increased porphyrin synthesis, deletion of the mitoferrin gene in mouse liver resulted in a decreased ability to convert protoporphyrin IX into haem, leading to protoporphyria, chronic hepatic damage, cholestasis and bridging fibrosis showing similar characteristics to that seen in human EPP.
It is important to consider that mechanisms other than porphyrin-induced liver injury may occur associated with disruption of enzymes of the haem and related pathway, perhaps as a consequence of mitochondria malfunction. Although mitochondrial failure is observed in mice with partial knockout of the hydroxymethylbilane synthase (HMBS) gene, no chronic studies appear to have been conducted that might explain the incidence of liver cancer in AIP patients in which high levels of non-porphyrin precursors of haem are implicated in toxicity.119 That oxidative processes alone can be tumorigenic is illustrated by the mitochondrial disruption, oxidative stress and subsequent liver tumors that are observed in mice with mutation of the frataxin gene which is involved in hepatic iron and heme metabolism.120 In a fumarylcetoacetate hydrolase (FAH) knockout mouse model of type 1 tyrosinemia (section 3) producing succinylacetone that inhibits ALAD, sustained activation of stress pathways were associated with hepatocellular damage, steatosis, oval cell proliferation and development of liver adenomas when the protective effect of nitrisinone was withdrawn.138 One possibility is that this is due to alkylating activity of tyrosine metabolites and related products.
6. An adverse outcome pathway for porphyria-induced liver cancer in rodents
An adverse outcome pathway (AOP) has been proposed for liver cancer induced by dioxin-like compounds through promotion following sustained activation of the AHR.56Fig. 8a summarizes this AOP developed by Becker et al.56 In this case the molecular initiating event (MIE) is binding and activation of the AHR but while the MIE is a necessary effect, it is not, on its own sufficient to produce the adverse outcome (AO). The downstream biological effects, termed key events (KE), within the proposed mode of action (MOA) for the development of liver cancers are similarly necessary but not in isolation, sufficient to provoke the final AO.121,122 This proposal suggests that following activation of the AHR, downstream KEs involve changes in the dynamics of cellular growth as a response to altered gene expression following AHR activation, observed as the development of hepatic foci, and decreases in the apoptotic rate both generally and especially within the foci, extensive liver toxicity described in the publication as “toxic hepatopathy”, and subsequent cellular proliferation and hyperplasia in several hepatic cell types. This progression of KEs then leads to the development of hepatocellular and cholangiocellular neoplasms. There is experimental evidence to support much of the proposed AOP but a critical review of the dose response and time relationships between the various key events and their subsequent KE would suggest that the sequence, although not necessarily the roles, of several of the KE may need to be evolved. Even with potent AHR ligands hepatic neoplasia still appears to only occur at relatively high dose levels when saturation of the AHR binding and activation would be expected to occur. Indeed in experimental situations in rodents, the dose–response for neoplasia correlates best with elevations in liver weight above a threshold.109,111,112 The causes of the liver weight induction with these AHR agonists are also unlikely to be a simple product of sustained AHR activation since the “toxic hepatopathy” is far more likely to be the main driver for the chronic regenerative hepatic hyperplasia that is implicit in the AOP. For those AHR agonists that induce porphyria, a main initiator of the “toxic hepatopathy” could be the porphyria that develops following dosing at high concentrations and the hepatotoxicity induced by the build-up of porphyrin. Its interference with biliary excretion are most likely major KEs that determine the evolution/promote the development of neoplasia in the rodent livers through chronic liver injury and the regenerative cell replication that ensues.123,124 Support for a potential role for hepatic porphyrin accumulation in the induction of neoplasia comes from the induction of rodent hepatic neoplasia by compounds that don't activate the AHR but that do induce porphyria, and the subsequent “toxic hepatopathy”, through inhibition of ferrochelatase. These compounds also induce CYPs, although there is no evidence of their activation of the AHR, but their commonality with the AHR agonists is the production of porphyria and the development of toxic hepatopathy. Therefore a revision of the Becker et al. AOP56 is presented below (Fig. 8b) which changes the order of some of the KEs but also introduces the key role of porphyrin in the subsequent induction of the hepatopathy and consequentially of all of the downstream events leading to hepatic neoplasia.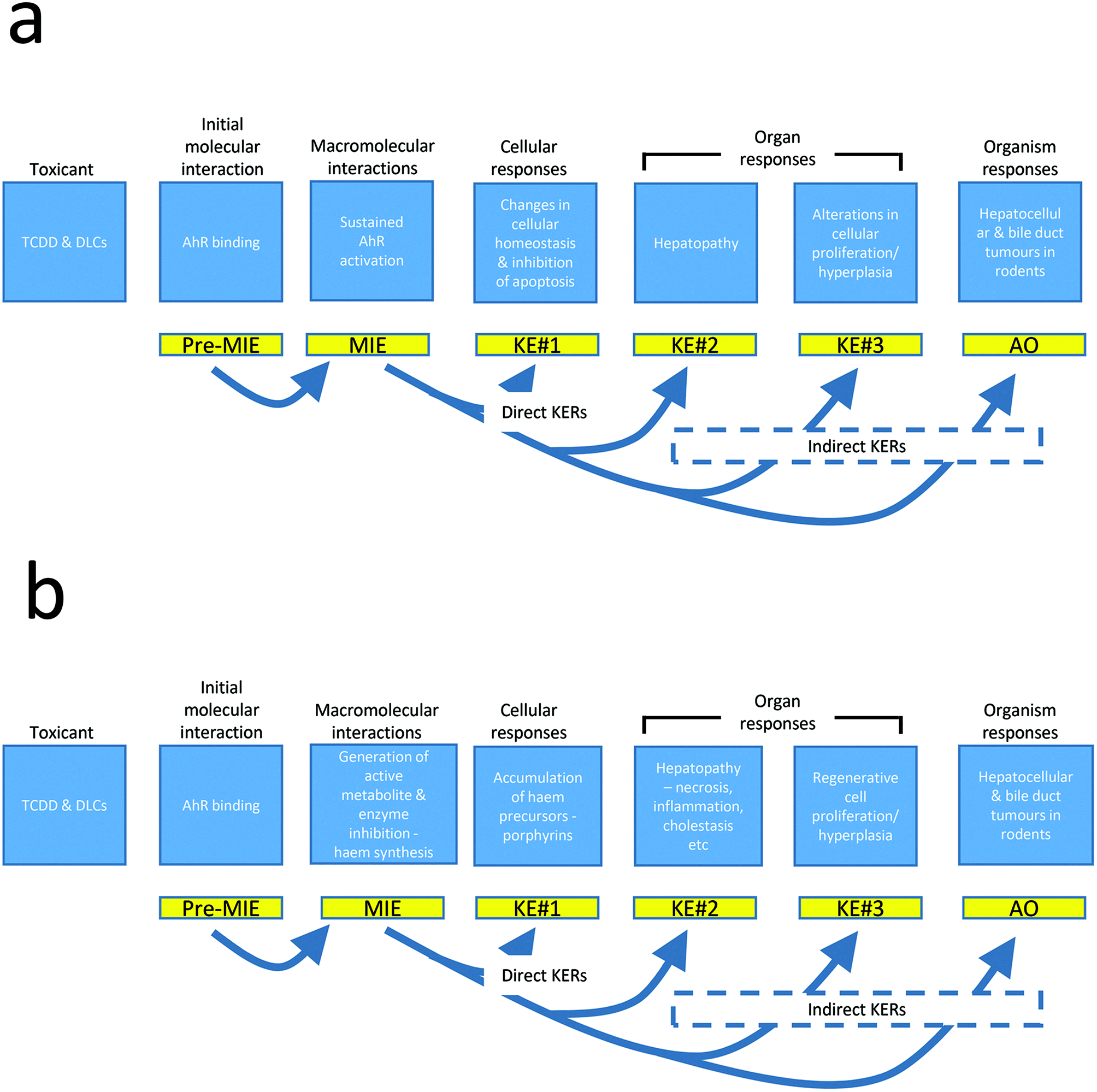 | ||
| Fig. 8 (a) An adverse outcome pathway analysis for the production of liver cancer with AHR ligands (redrawn from Becker et al. 2015).56 (b) An alternative AOP for the production of liver cancer in rodents via the hepatic accumulation of porphyrins. | ||
7. Summary
Some chemicals, drugs and pesticides are known to interfere in experimental animals with hepatic haem synthesis causing porphyria. With some clinical human porphyrias there is a known association with increased incidences of hepatocellular cancer. Hepatic porphyrin accumulation caused in rodents may occur either by mechanisms that produce oxidized haem precursors which act as inhibitors of enzymes in the pathway or by metabolism of the chemical that by a side reaction produces an inhibitor of ferrochelatase, from the haem of cytochrome P450. In both mechanisms there is evidence that under chronic conditions the accumulation of porphyrins, mostly either uroporphyrins or protoporphyrin IX may cause liver injury and aid subsequent liver tumour formation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Preliminary presentation on this topic took place to a HESI Workshop U.S. EPA, 2004.125References
- W. E. Daniell, H. L. Stockbridge, R. F. Labbe, J. S. Woods, K. E. Anderson, D. M. Bissell, J. R. Bloomer, R. D. Ellefson, M. R. Moore, C. A. Pierach, W. E. Schreiber, A. Tefferi and G. M. Franklin, Environmental chemical exposures and disturbances of heme synthesis, Environ. Health Perspect., 1997, 105, 37–53 CrossRef PubMed.
- F. De Matteis and G. S. Marks, Cytochrome P450 and its interactions with the heme biosynthetic pathway, Can. J. Physiol. Pharmacol., 1996, 74, 1–8 CrossRef PubMed.
- A. G. Smith and G. H. Elder, Complex gene-chemical interactions: hepatic uroporphyria as a paradigm, Chem. Research Toxicol., 2010, 23, 712–723 CrossRef PubMed.
- V. Vagany and A. G. Smith, Complicity of haem in some adverse drug-reactions, Toxicol. Res., 2015, 1128–1142 RSC.
- Z. Karim, S. Lyoumi, G. Nicolas, J. C. Deybach, L. Gouya and H. Puy, Porphyrias: A 2015 update, Clin. Res. Hepatol. Gastroenterol., 2015, 39, 412–425 CrossRef PubMed.
- H. Puy, L. Gouya and J. C. Deybach, Porphyrias, Lancet, 2010, 375, 924–937 CrossRef.
- J. Berman and A. Braun, Incidence of hepatoma in porphyria cutanea tarda, Rev. Czech. Med., 1962, 8, 290–295 Search PubMed.
- N. O. Bengtsson and L. Hardell, Porphyrias, porphyrins and hepatocellular cancer, Br. J. Cancer, 1986, 54, 115–117 CrossRef PubMed.
- F. Lithner and L. Wetterberg, Hepatocellular carcinoma in patients with acute intermittent porphyria, Acta Med. Scand., 1984, 21, 271–274 Search PubMed.
- V. Kordac, Frequency of occurrence of hepatocellular carcinoma in patients with porphyria cutanea tarda in long-term follow-up, Neoplasia, 1972, 19, 135–139 Search PubMed.
- Z. F. Soonawalla, T. Orug, M. N. Badminton, G. H. Elder, J. M. Rhodes, S. R. Bramhall and E. Elias, Liver transplantation as a cure for acute intermittent porphyria, Lancet, 2004, 363, 705–706 CrossRef.
- D. M. Bissell, J. C. Lai, R. K. Meister and P. D. Blanc, Role of delta-aminolevulinic acid in the symptoms of acute porphyria, Am. J. Med., 2015, 128, 313–317 CrossRef PubMed.
- D. M. Bissell and B. Wang, Acute hepatic porphyria, J. Clin. Transl. Hepatol., 2015, 3, 17–26 CrossRef PubMed.
- R. Kauppinen and P. Mustajoki, Acute hepatic porphyria and hepatocellular carcinoma, Br. J. Cancer, 1988, 57, 117–120 CrossRef PubMed.
- M. J. Tidman, E. M. Higgins, G. H. Elder and D. M. MacDonald, Variegate porphyria associated with hepatocellular carcinoma, Br. J. Dermatol., 1989, 121, 503–505 CrossRef PubMed.
- C. Andant, H. Puy, C. Bogard, J. Faivre, J. C. Soule, Y. Nordmann and J. C. Deybach, Hepatocellular carcinoma in patients with acute hepatic porphyria: frequency of occurrence and related factors, J. Hepatol., 2000, 32, 933–939 CrossRef PubMed.
- X. Schneider-Yin, A. M. Van Tuyll Van Serooskerken, P. Went, W. Tyblewski, P. Poblete-Gutiérrez, E. I. Minder and J. Frank, Hepatocellular carcinoma in variegate porphyria: A serious complication, Acta Derm.-Venereol., 2010, 90, 512–515 CrossRef PubMed.
- E. Innala and C. Andersson, Screening for hepatocellular carcinoma in acute intermittent porphyria: a 15-year follow-up in northern Sweden, J. Intern. Med., 2011, 26, 538–545 CrossRef PubMed.
- E. Sardh, S. Wahlin, M. Bjornstedt, P. Harper and D. E. Andersson, High risk of primary liver cancer in a cohort of 179 patients with acute hepatic porphyria, J. Inherited Metab. Dis., 2013, 1063–1071 CrossRef PubMed.
- C. M. Baravelli, S. Sandberg, A. K. Aarsand, R. M. Nilsen and M. C. Tollanes, Acute hepatic porphyria and cancer risk: a nationwide cohort study, J. Intern. Med., 2017, 282, 229–240 CrossRef PubMed.
- X. Schneider-Yin, A. M. van Tuyll van Serooskerken, M. Siegesmund, P. Went, J. Barman-Aksozen, R. S. Bladergroen, P. Komminoth, R. H. Cloots, V. J. Winnepenninckx, A. zur Hausen, M. Weber, A. Driessen, P. Poblete-Gutierrez, P. Bauer, C. Schroeder, M. van Geel, E. I. Minder and J. Frank, Biallelic inactivation of protoporphyrinogen oxidase and hydroxymethylbilane synthase is associated with liver cancer in acute porphyrias, J. Hepatol., 2015, 62, 734–738 CrossRef PubMed.
- J. Onuki, P. C. Teixeira, M. H. Medeiros, D. Dornemann, T. Douki, J. Cadet and P. Di Mascio, Is 5-aminolevulinic acid involved in the hepatocellular carcinogenesis of acute intermittent porphyria?, Cell. Mol. Biol., 2002, 48, 17–26 Search PubMed.
- J. C. Deybach and H. Puy, Hepatocellular carcinoma without cirrhosis: think acute hepatic porphyrias and vice versa, J. Intern. Med., 2011, 269, 521–524 CrossRef PubMed.
- A. G. Roberts, S. D. Whatley, R. R. Morgan, M. Worwood and G. H. Elder, Increased frequency of the haemochromatosis Cys282Tyr mutation in sporadic porphyria cutanea tarda, Lancet, 1997, 349, 321–323 CrossRef.
- J. M. Cortes, H. Oliva, F. J. Paradinas and C. Hernandez-Guio, The pathology of the liver in porphyria cutanea tarda, Histopathology, 1980, 4, 471–485 CrossRef PubMed.
- H. Salata, J. M. Cortes, R. Enriquez de Salamanca, H. Oliva, A. Castro, E. Kusak, V. Carreno and C. Hernandez Guio, Porphyria cutanea tarda and hepatocellular carcinoma. Frequency of occurrence and related factors, J. Hepatol., 1985, 1, 477–487 CrossRef PubMed.
- P. D. Siersema, F. J. ten Kate, P. G. Mulder and J. H. Wilson, Hepatocellular carcinoma in porphyria cutanea tarda: frequency and factors related to its occurrence, Liver, 1992, 12, 56–61 CrossRef PubMed.
- G. C. Topi, L. D'Alessandro Gandolfo, D. Griso and S. Morini, Porphyria cutanea tarda and hepatocellular carcinoma, Int. J. Biochem., 1980, 12, 883–885 CrossRef PubMed.
- J. P. Gisbert, L. Garcia-Buey, A. Alonso, S. Rubio, A. Hernández, J. M. Pajares, A. García-Díez and R. Moreno-Otero, Hepatocellular carcinoma risk in patients with porphyria cutanea tarda, Eur. J. Gastroenterol. Hepatol., 2004, 16, 689–692 CrossRef PubMed.
- A. L. Fracanzani, E. Taioli, M. Sampietro, E. Fatta, C. Bertelli, G. Fiorelli and S. Fargion, Liver cancer risk is increased in patients with porphyria cutanea tarda in comparison to matched control patients with chronic liver disease, J. Hepatol., 2001, 35, 498–503 CrossRef PubMed.
- V. Kordac, P. Martasek, V. Zoubek and M. Kalab, Porphyria cutanea-tarda - manifestation and therapy, Ann. N. Y. Acad. Sci., 1987, 514, 335–336 CrossRef.
- R. L. Lindberg, C. Porcher, B. Grandchamp, B. Ledermann, K. Burki, S. Brandner, A. Aguzzi and U. A. Meyer, Porphobilinogen deaminase deficiency in mice causes a neuropathy resembling that of human hepatic porphyria, Nat. Genet., 1996, 12, 195–199 CrossRef PubMed.
- C. Handschin, J. Lin, J. Rhee, A. K. Peyer, S. Chin, P. H. Wu, U. A. Meyer and B. M. Spiegelman, Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha, Cell, 2005, 122, 505–515 CrossRef PubMed.
- J. E. Francis and A. G. Smith, Oxidation of uroporphyrinogens by hydroxyl radicals. Evidence for nonporphyrin products as potential inhibitors of uroporphyrinogen decarboxylase, FEBS Lett., 1988, 233, 311–314 CrossRef PubMed.
- J. D. Phillips, H. A. Bergonia, C. A. Reilly, M. R. Franklin and J. P. Kushner, A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5079–5084 CrossRef PubMed.
- M. Van den Berg, L. S. Birnbaum, M. Denison, M. De Vito, W. Farland, M. Feeley, H. Fiedler, H. Hakansson, A. Hanberg, L. Haws, M. Rose, S. Safe, D. Schrenk, C. Tohyama, A. Tritscher, J. Tuomisto, M. Tysklind, N. Walker and R. E. Peterson, The 2005 World Health Organization reevaluation of human and mammalian toxic equivalency factors for dioxins and dioxin-like compounds, Toxicol. Sci., 2006, 93, 223–241 CrossRef PubMed.
- A. P. van Birgelen, Hexachlorobenzene as a possible major contributor to the dioxin activity of human milk, Environ. Health Perspect., 1998, 106, 683–688 CrossRef PubMed.
- M. E. Hahn, T. A. Gasiewicz, P. Linko and J. A. Goldstein, The role of the Ah locus in hexachlorobenzene-induced porphyria, Studies in congenic C57BL/6J mice, Biochem. J., 1988, 254, 245–254 CrossRef PubMed.
- M. E. Hahn, J. A. Goldstein, P. Linko and T. A. Gasiewicz, Interaction of hexachlorobenzene with the receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin in vitro and in vivo. Evidence that hexachlorobenzene is a weak Ah receptor agonist, Arch. Biochem. Biophys., 1989, 270, 344–355 CrossRef PubMed.
- R. A. Akhtar and A. G. Smith, Chromosomal linkage analysis of porphyria in mice induced by hexachlorobenzene-iron synergism: a model of sporadic porphyria cutanea tarda, Pharmacogenetics, 1998, 8, 485–494 CrossRef PubMed.
- M. D. Stonard, Mixed type hepatic microsomal enzyme induction by hexachlorobenzene, Biochem. Pharmacol., 1975, 24, 1959–1963 CrossRef PubMed.
- M. D. Stonard and J. B. Greig, Different patterns of hepatic microsomal-enzyme activity produced by administration of pure hexachlorobiphenyl isomers and hexachlorobenzene, Chem.-Biol. Interact., 1976, 15, 365–379 CrossRef PubMed.
- A. G. Smith, J. E. Francis and I. Bird, Distinction between octachlorostyrene and hexachlorobenzene in their potentials to induce ethoxyphenoxazone deethylase and cause porphyria in rats and mice, J. Biochem. Toxicol., 1986, 1, 105–117 CrossRef.
- L. Cantoni, M. Rizzardini, M. T. Tacconi and A. Graziani, Comparison of hexachlorobenzene-induced alterations of microsomal membrane composition and monooxygenase activity in male and female rats, Toxicology, 1987, 45, 291–305 CrossRef PubMed.
- D. L. Grant, F. Iverson, G. V. Hatina and D. C. Villeneuve, Effects of hexachlorobenzene on liver porphyrin levels and microsomal enzymes in the rat, Environ. Physiol. Biochem., 1974, 4, 159–165 Search PubMed.
- A. G. Smith, J. E. Francis, J. A. Green, J. B. Greig, C. R. Wolf and M. M. Manson, Sex-linked hepatic uroporphyria and the induction of cytochromes P450IA in rats caused by hexachlorobenzene and polyhalogenated biphenyls, Biochem. Pharmacol., 1990, 40, 2059–2068 CrossRef PubMed.
- A. G. Smith, J. E. Francis, D. Dinsdale, M. M. Manson and J. R. Cabral, Hepatocarcinogenicity of hexachlorobenzene in rats and the sex difference in hepatic iron status and development of porphyria, Carcinogenesis, 1985, 6, 631–636 CrossRef PubMed.
- D. L. Arnold, C. A. Moodie, S. M. Charbonneau, H. C. Grice, P. F. McGuire, F. R. Bryce, B. T. Collins, Z. Z. Zawidzka, D. R. Krewski and E. A. Nera, et al., Long-term toxicity of hexachlorobenzene in the rat and the effect of dietary vitamin A, Food Chem. Toxicol., 1985, 23, 779–793 CrossRef PubMed.
- E. Erturk, R. W. Lambrecht, H. A. Peters, D. J. Cripps, A. Gocmen, C. R. Morris and G. T. Bryan, Oncogenicity of hexachlorobenzene, IARC Sci. Publ., 1986, 77, 417–423 Search PubMed.
- D. H. Norback and R. H. Weltman, Polychlorinated biphenyl induction of hepatocellular carcinoma in the Sprague-Dawley rat, Environ. Health Perspect., 1985, 60, 97–105 CrossRef PubMed.
- R. D. Kimbrough, V. W. Burse and J. A. Liddle, Persistent liver lesions in rats after a single oral dose of polybrominated biphenyls (firemaster FF-1) and concomitant PBB tissue levels, Environ. Health Perspect., 1978, 23, 265–273 CrossRef PubMed.
- R. D. Kimbrough, D. F. Groce, M. P. Korver and V. W. Burse, Induction of liver tumors in female Sherman strain rats by polybrominated biphenyls, J. Natl. Cancer Inst., 1981, 66, 535–542 Search PubMed.
- R. J. Kociba, D. G. Keyes, J. E. Beyer, R. M. Carreon, C. E. Wade, D. A. Dittenber, R. P. Kalnins, L. E. Frauson, C. N. Park, S. D. Barnard, R. A. Hummel and C. G. Humiston, Results of a two-year chronic toxicity and oncogenicity study of 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats, Toxicol. Appl. Pharmacol., 1978, 46, 279–303 CrossRef PubMed.
- P. Carthew and A. G. Smith, Pathological mechanisms of hepatic tumour formation in rats exposed chronically to dietary hexachlorobenzene, J. Appl. Toxicol., 1994, 14, 447–452 CrossRef PubMed.
- R. A. Budinsky, D. Schrenk, T. Simon, M. Van den Berg, J. F. Reichard, J. B. Silkworth, L. L. Aylward, A. Brix, T. Gasiewicz, N. Kaminski, G. Perdew, T. B. Starr, N. J. Walker and J. C. Rowlands, Mode of action and dose-response framework analysis for receptor-mediated toxicity: The aryl hydrocarbon receptor as a case study, Crit. Rev. Toxicol., 2014, 44, 83–119 CrossRef PubMed.
- R. A. Becker, G. Patlewicz, T. W. Simon, J. C. Rowlands and R. A. Budinsky, The adverse outcome pathway for rodent liver tumor promotion by sustained activation of the aryl hydrocarbon receptor, Regul. Toxicol. Pharmacol., 2015, 73, 172–190 CrossRef PubMed.
- G. D. Kennedy, M. Nukaya, S. M. Moran, E. Glover, S. Weinberg, S. Balbo, S. S. Hecht, H. C. Pitot, N. R. Drinkwater and C. A. Bradfield, Liver tumor promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin is dependent on the aryl hydrocarbon receptor and TNF/IL-1 receptors, Toxicol. Sci., 2014, 140, 135–143 CrossRef PubMed.
- F. P. Stewart, M. M. Manson, J. R. Cabral and A. G. Smith, Hexachlorobenzene as a promoter of diethylnitrosamine-initiated hepatocarcinogenesis in rats and comparison with induction of porphyria, Carcinogenesis, 1989, 10, 1225–1230 CrossRef PubMed.
- A. G. Smith, J. R. Cabral and F. De Matteis, A difference between two strains of rats in their liver non-haem iron content and in their response to the porphyrogenic effect of hexachlorobenzene, Chem.-Biol. Interact., 1979, 27, 353–363 CrossRef PubMed.
- A. G. Smith, P. Carthew, J. E. Francis, J. R. Cabral and M. M. Manson, Enhancement by iron of hepatic neoplasia in rats caused by hexachlorobenzene, Carcinogenesis, 1993, 14, 1381–1387 CrossRef PubMed.
- A. G. Smith and J. E. Francis, Synergism of iron and hexachlorobenzene inhibits hepatic uroporphyrinogen decarboxylase in inbred mice, Biochem. J., 1983, 214, 909–913 CrossRef PubMed.
- J. B. Greig, J. E. Francis, S. J. Kay, D. P. Lovell and A. G. Smith, Incomplete correlation of 2,3,7,8-tetrachlorodibenzo-p-dioxin hepatotoxicity with Ah phenotype in mice, Toxicol. Appl. Pharmacol., 1984, 74, 17–25 CrossRef PubMed.
- S. W. Robinson, B. Clothier, R. A. Akhtar, A. L. Yang, I. Latour, C. Van Ijperen, M. F. Festing and A. G. Smith, Non-ahr gene susceptibility loci for porphyria and liver injury induced by the interaction of ‘dioxin’ with iron overload in mice, Mol. Pharmacol., 2002, 61, 674–681 CrossRef PubMed.
- A. G. Smith, J. R. Cabral, P. Carthew, J. E. Francis and M. M. Manson, Carcinogenicity of iron in conjunction with a chlorinated environmental chemical, hexachlorobenzene, in C57BL/10ScSn mice, Int. J. Cancer, 1989, 43, 492–496 CrossRef PubMed.
- A. G. Smith, J. E. Francis and P. Carthew, Iron as a synergist for hepatocellular carcinoma induced by polychlorinated biphenyls in Ah-responsive C57BL/10ScSn mice, Carcinogenesis, 1990, 11, 437–444 CrossRef PubMed.
- A. G. Smith, B. Clothier, P. Carthew, N. L. Childs, P. R. Sinclair, D. W. Nebert and T. P. Dalton, Protection of the Cyp1a2(-/-) null mouse against uroporphyria and hepatic injury following exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin, Toxicol. Appl. Pharmacol., 2001, 173, 89–98 CrossRef PubMed.
- P. R. Sinclair, N. Gorman, H. S. Walton, W. J. Bement, T. P. Dalton, J. F. Sinclair, A. G. Smith and D. W. Nebert, CYP1A2 is essential in murine uroporphyria caused by hexachlorobenzene and iron, Toxicol. Appl. Pharmacol., 2000, 162, 60–67 CrossRef PubMed.
- P. Greaves, B. Clothier, R. Davies, F. M. Higginson, R. E. Edwards, T. P. Dalton, D. W. Nebert and A. G. Smith, Uroporphyria and hepatic carcinogenesis induced by polychlorinated biphenyls-iron interaction: absence in the Cyp1a2(-/-) knockout mouse, Biochem. Biophys. Res. Commun., 2005, 331, 147–152 CrossRef PubMed.
- A. G. Smith, P. Carthew, J. E. Francis and K. Ingebrigtsen, Influence of iron on the induction of hepatic tumors and porphyria by octachlorostyrene in C57BL/10ScSn mice, Cancer Lett., 1994, 81, 145–150 CrossRef PubMed.
- S. Madra, J. Styles and A. G. Smith, Perturbation of hepatocyte nuclear populations induced by iron and polychlorinated biphenyls in C57BL/10ScSn mice during carcinogenesis, Carcinogenesis, 1995, 16, 719–727 CrossRef PubMed.
- P. C. Rumsby, J. G. Evans, H. E. Phillimore, P. Carthew and A. G. Smith, Search for Ha-ras codon 61 mutations in liver tumours caused by hexachlorobenzene and Aroclor 1254 in C57BL/10ScSn mice with iron overload, Carcinogenesis, 1992, 13, 1917–1920 CrossRef PubMed.
- S. P. Faux, J. E. Francis, A. G. Smith and J. K. Chipman, Induction of 8-hydroxydeoxyguanosine in Ah-responsive mouse liver by iron and Aroclor 1254, Carcinogenesis, 1992, 13, 247–250 CrossRef PubMed.
- R. Davies, B. Clothier and A. G. Smith, Mutation frequency in the lacI gene of liver DNA from lambda/lacI transgenic mice following the interaction of PCBs with iron causing hepatic cancer and porphyria, Mutagenesis, 2000, 15, 379–383 CrossRef PubMed.
- F. G. Dayan and S. O. Duke, Protoporphyrinogen oxidase-inhibiting herbicides, in Hayes Handbook of Pesticide Toxicology, ed. R. Krieger, Academic Press, 2010, ch. 81, vol. 2, pp. 1733–1751 Search PubMed.
- J. Krijt, M. Vokurka, J. Sanitrak, V. Janousek, L. van Holsteijn and B. J. Blaauboer, Effect of the protoporphyrinogen oxidase-inhibiting herbicide fomesafen on liver uroporphyrin and heptacarboxylic porphyrin in two mouse strains, Food Chem. Toxicol., 1994, 32, 641–650 CrossRef PubMed.
- J. Krijt, O. Pšenák, M. Vokurka, A. Chlumská and F. Fakan, Experimental hepatic uroporphyria induced by the diphenyl-ether herbicide fomesafen in male DBA/2 mice, Toxicol. Appl. Pharmacol., 2003, 189, 28–38 CrossRef PubMed.
- J. Krijt, P. Stranska, J. Sanitrak, A. Chlumska and F. Fakan, Liver preneoplastic changes in mice treated with the herbicide fomesafen, Hum. Exp. Toxicol., 1999, 18, 338–344 Search PubMed.
- M. Sachar, K. E. Anderson and X. Ma, Protoporphyrin IX: the good, the bad, and the ugly, J. Pharmacol. Exp. Ther., 2016, 356, 267–275 CrossRef PubMed.
- F. De Matteis, A. H. Gibbs, A. H. Jackson and S. Weerasinghe, Conversion of liver haem into N-substituted porphyrins or green pigments. Nature of the substituent at the pyrrole nitrogen atom, FEBS Lett., 1980, 119, 109–112 CrossRef PubMed.
- J. T. Gamble, K. Nakatsu and G. S. Marks, Comparison of rat and human cytochrome P450 (CYP) sources of N-alkylprotoporphyrin IX. Formation after interaction with porphyrinogenic xenobiotics: studies with cDNA-expressed single CYP enzymes, Xenobiotica, 2002, 32, 997–1006 CrossRef PubMed.
- J. T. Gamble, S. G. Wong, H. A. Dailey and G. S. Marks, Use of recombinant human ferrochelatase as a sensitive bioassay for N-alkylprotoporphyrin IX formed after interaction of porphyrinogenic xenobiotics with rat liver microsomes, Can. J. Physiol. Pharmacol., 2000, 78, 578–581 CrossRef PubMed.
- K. Nakao, O. Wada, F. Takaku, S. Sassa, Y. Yano and G. Urata, The origin of the increased protoporphyrin in erythrocytes of mice with experimentally induced porphyria, J. Lab. Clin. Med., 1967, 70, 923–932 Search PubMed.
- A. Holley, L. J. King, A. H. Gibbs and F. De Matteis, Strain and sex differences in the response of mice to drugs that induce protoporphyria: role of porphyrin biosynthesis and removal, J. Biochem. Toxicol., 1990, 5, 175–182 CrossRef.
- F. De Matteis, A. H. Gibbs, P. B. Farmer and J. H. Lamb, Liver production of N-alkylated porphyrins caused in mice by treatment with substituted dihydropyridines. Evidence that the alkyl group on the pyrrole nitrogen atom originates from the drug, FEBS Lett., 1981, 129, 328–331 CrossRef PubMed.
- F. De Matteis and A. H. Gibbs, Stimulation of the pathway of porphyrin synthesis in the liver of rats and mice by griseofulvin, 3,5-diethoxycarbonyl-1,4-dihydrocollidine and related drugs: evidence for two basically different mechanisms, Biochem. J., 1975, 146, 285–287 CrossRef PubMed.
- T. W. Gant, P. R. Baus, B. Clothier, J. Riley, R. Davies, D. J. Judah, R. E. Edwards, E. George, P. Greaves and A. G. Smith, Gene expression profiles associated with inflammation, fibrosis, and cholestasis in mouse liver after griseofulvin, Environ. Health Perspect., 2003, 111, 847–853 Search PubMed.
- R. Stejskal, M. Itabashi, J. Stanek and Z. Hruban, Experimental porphyria induced by (3-(2,4,6-trimethylphenyl)-thioethyl)-4 methylsydnone, Virchows Arch. B, 1975, 18, 83–100 Search PubMed.
- A. M. Brady, R. L. Hasmall, B. M. Elcombe, E. A. Lock and F. De Matteis, Effects of 2-[1-(ethoxyimino)propyl]-3-hydroxy-5-(2,4,6-trimethylphenyl) cyclohex-2-enone on hepatic haem biosynthesis: species differences in hepatic porphyria, Toxicology, 1993, 84, 199–215 CrossRef PubMed.
- Y. Frater, A. Brady, E. A. Lock and F. De Matteis, Formation of N-methyl protoporphyrin in chemically-induced protoporphyria. Studies with a novel porphyrogenic agent, Arch. Toxicol., 1993, 67, 179–185 CrossRef PubMed.
- F. De Matteis, A. H. Gibbs and A. G. Smith, Inhibition of protohaem ferro-lyase by N-substituted porphyrins. Structural requirements for the inhibitory effect, Biochem. J., 1980, 189, 645–648 CrossRef PubMed.
- F. De Matteis, A. H. Gibbs, L. Cantoni and J. Francis, Substrate-dependent irreversible inactivation of cytochrome P-450: conversion of its haem moiety into modified porphyrins, Ciba Found. Symp., 1980, 76, 119–139 Search PubMed.
- I. N. White, Destruction of liver haem by norethindrone. Conversion into green pigments, Biochem. J., 1981, 196, 575–583 CrossRef PubMed.
- S. Knasmuller, W. Parzefall, C. Helma, F. Kassie, S. Ecker and R. Schulte-Hermann, Toxic effects of griseofulvin: disease models, mechanisms, and risk assessment, Crit. Rev. Toxicol., 1997, 27, 495–537 CrossRef PubMed.
- J. M. Nicolas, H. Chanteux, V. Mancel, G. M. Dubin, B. Gerin, L. Staelens, O. Depelchin and S. Kervyn, N-alkylprotoporphyrin formation and hepatic porphyria in dogs after administration of a new antiepileptic drug candidate: Mechanism and species specificity, Toxicol. Sci., 2014, 141, 353–364 CrossRef PubMed.
- F. De Matteis, A. J. Donnelly and W. J. Runge, The effects of prolonged administration of griseofulvin in mice with reference to sex differences, Cancer Res., 1966, 26, 721–726 Search PubMed.
- A. Matilla and E. A. Molland, A light and electron microscopic study of the liver in case of erythrohepatic protoporphyria and in griseofulvin-induced porphyria in mice, J. Clin. Pathol., 1974, 27, 698–709 CrossRef PubMed.
- F. Gschnait, K. Konrad, H. Honigsmann, H. Denk and K. Wolff, Mouse model for protoporphyria. I. The liver and hepatic protoporphyrin crystals, J. Invest. Dermatol., 1975, 65, 290–299 CrossRef PubMed.
- S. W. Choi, J. H. Han, K. T. Lim, H. M. Cho, K. W. Chung, H. S. Sun, D. H. Park, B. S. Kim and E. J. Seo, Effect of ursodeoxycholic acid on experimental hepatic porphyria induced by griseofulvin, J. Korean Med. Sci., 1991, 6, 146–156 CrossRef PubMed.
- B. D. Pauli and S. W. Kennedy, Hepatic porphyria induced by the herbicide tralkoxydim in small mammals is species-specific, Environ. Toxicol. Chem., 2005, 24, 450–456 CrossRef PubMed.
- EFSA Report, Conclusion on the peer review of the pesticide risk assessment of the active substance Tralkoxydim, EFSA J., 2008, 139, 1–78 Search PubMed.
- F. de Matteis, C. Hollands, A. H. Gibbs, N. de Sa and M. Rizzardini, Inactivation of cytochrome P-450 and production of N-alkylated porphyrins caused in isolated hepatocytes by substituted dihydropyridines. Structural requirements for loss of haem and alkylation of the pyrrole nitrogen atom, FEBS Lett., 1982, 145, 87–92 CrossRef PubMed.
- A. M. Brady and E. A. Lock, Inhibition of ferrochelatase and accumulation of porphyrins in mouse hepatocyte cultures exposed to porphyrinogenic chemicals, Arch. Toxicol., 1992, 66, 175–181 CrossRef PubMed.
- W. Y. Kao, C. W. Su, Y. S. Huang, Y. C. Chou, Y. C. Chen, W. H. Chung, M. C. Hou, H. C. Lin, F. Y. Lee and J. C. Wu, Risk of oral antifungal agent-induced liver injury in Taiwanese, Br. J. Clin. Pharmacol., 2014, 77, 180–189 CrossRef PubMed.
- C. Rimington, P. N. Morgan, K. Nicholls, J. D. Everall and R. R. Davies, Griseofulvin administration and porphyrin metabolism. A survey, Lancet, 1963, 2, 318–322 CrossRef.
- A. Berman and R. L. Franklin, Precipitation of acute intermittent porphyria by griseofulvin therapy, J. Am. Med. Assoc., 1965, 192, 1005–1007 CrossRef.
- C. Martinez Mdel, S. G. Afonso, R. P. Meiss, A. M. Buzaleh and A. Batlle, Hepatic damage and oxidative stress induced by griseofulvin in mice, Cell Mol. Biol., 2009, 55, 127–139 Search PubMed.
- D. E. Hatoff and W. G. Hardison, Bile acids modify alkaline phosphatase induction and bile secretion pressure after bile duct obstruction in the rat, Gastroenterology, 1981, 80, 666–672 Search PubMed.
- E. Weston-Hurst and G. E. Paget, Protoporphyrin, cirrhosis and hepatomata in the livers of mice given griseofulvin, Br. J. Dermatol., 1963, 75, 105–112 CrossRef.
- M. Rustia and P. Shubik, Thyroid tumours in rats and hepatomas in mice after griseofulvin treatment, Br. J. Cancer, 1978, 38, 237–249 CrossRef PubMed.
- C. F. Polo, A. M. Buzaleh, E. S. Vazquez, S. G. Afonso, N. M. Navone and A. M. Batlle, Griseofulvin-induced hepatopathy due to abnormalities in heme pathway, Gen. Pharmacol., 1997, 29, 207–210 CrossRef PubMed.
- T. Shimoyama and S. Nonaka, Biochemical studies on griseofulvin-induced protoporphyria, Ann. N. Y. Acad. Sci., 1987, 514, 160–169 CrossRef PubMed.
- T. Shimoyama, S. Nonaka, T. Honda, F. Murayama, T. Ohgami and H. Yoshida, A biochemical study of experimental porphyria. I. The influence of griseofulvin at various concentrations on porphyrin metabolism, J. Dermatol., 1984, 11, 49–55 CrossRef PubMed.
- S. Tutois, X. Montagutelli, V. Da Silva, H. Jouault, P. Rouyer-Fessard, K. Leroy-Viard, J. L. Guenet, Y. Nordmann, Y. Beuzard and J. C. Deybach, Erythropoietic protoporphyria in the house mouse. A recessive inherited ferrochelatase deficiency with anemia, photosensitivity, and liver disease, J. Clin. Invest., 1991, 88, 1730–1736 CrossRef PubMed.
- R. Davies, A. Schuurman, C. R. Barker, B. Clothier, T. Chernova, F. M. Higginson, D. J. Judah, D. Dinsdale, R. E. Edwards, P. Greaves, T. W. Gant and A. G. Smith, Hepatic gene expression in protoporphyic Fech mice is associated with cholestatic injury but not a marked depletion of the heme regulatory pool, Am. J. Pathol., 2005, 166, 1041–1053 CrossRef PubMed.
- L. Libbrecht, L. Meerman, F. Kuipers, T. Roskams, V. Desmet and P. Jansen, Liver pathology and hepatocarcinogenesis in a long-term mouse model of erythropoietic protoporphyria, J. Pathol., 2003, 199, 191–200 CrossRef PubMed.
- J. Barman-Aksozen, C. W. P. V. B. Bansode, F. Koentgen, J. Trub, P. Pelczar, P. Cinelli, X. Schneider-Yin, D. Schumperli and E. I. Minder, Modeling the ferrochelatase c.315–48C modifier mutation for erythropoietic protoporphyria (EPP) in mice, Dis. Models & Mech., 2017, 10, 225–233 Search PubMed.
- M. Lecha, H. Puy and J. C. Deybach, Erythropoietic protoporphyria, Orphanet. J. Rare Dis., 2009, 4, 19 CrossRef PubMed.
- M. B. Troadec, D. Warner, J. Wallace, K. Thomas, G. J. Spangrude, J. Phillips, O. Khalimonchuk, B. H. Paw, D. M. Ward and J. Kaplan, Targeted deletion of the mouse Mitoferrin1 gene: from anemia to protoporphyria, Blood, 2011, 117, 5494–5502 CrossRef PubMed.
- C. Homedan, J. Laafi, C. Schmitt, N. Gueguen, T. Lefebvre, Z. Karim, V. Desquiret-Dumas, C. Wetterwald, J. C. Deybach, L. Gouya, H. Puy, P. Reynier and Y. Malthiery, Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model, Int. J. Biochem. Cell Biol., 2014, 51, 93–101 CrossRef PubMed.
- R. Thierbach, T. J. Schulz, F. Isken, A. Voigt, B. Mietzner, G. Drewes, J. C. von Kleist-Retzow, R. J. Wiesner, M. A. Magnuson, H. Puccio, A. F. Pfeiffer, P. Steinberg and M. Ristow, Targeted disruption of hepatic frataxin expression causes impaired mitochondrial function, decreased life span and tumor growth in mice, Hum. Mol. Genet., 2005, 14, 3857–3864 CrossRef PubMed.
- E. Julien, A. R. Boobis and S. S. Olin, The Key Events Dose-Response Framework: a cross-disciplinary mode-of-action based approach to examining dose-response and thresholds, Crit. Rev. Food Sci. Nutr., 2009, 49, 682–689 CrossRef PubMed.
- T. W. Simon, S. S. Simons Jr., R. J. Preston, A. R. Boobis, S. M. Cohen, N. G. Doerrer, P. A. Fenner-Crisp, T. S. McMullin, C. A. McQueen and J. C. Rowlands, The use of mode of action information in risk assessment: quantitative key events/dose-response framework for modeling the dose-response for key events, Crit. Rev. Toxicol., 2014, 44(Suppl. 3), 17–43 CrossRef PubMed.
- S. M. Cohen and L. B. Ellwein, Cell proliferation in carcinogenesis, Science, 1990, 249, 1007–1011 Search PubMed.
- J. R. Foster, The role of cell proliferation in chemically induced carcinogenesis, J. Comp. Pathol., 1997, 116, 113–144 CrossRef PubMed.
- M. P. Holsapple, H. C. Pitot, S. M. Cohen, A. R. Boobis, J. E. Klaunig, T. Pastoor, V. L. Dellarco and Y. P. Dragan, Mode of action in relevance of rodent liver tumors to human cancer risk, Toxicol. Sci., 2006, 89, 51–56 CrossRef PubMed.
- L. Rossi, M. Ravera, G. Repetti and L. Santi, Long-term administration of DDT or phenobarbital-Na in Wistar rats, Int. J. Cancer, 1977, 19, 179–185 CrossRef PubMed.
- P. Greaves, R. Goonetilleke, G. Nunn, J. Topham and T. Orton, Two-year carcinogenicity study of tamoxifen in Alderley Park Wistar-derived rats, Cancer Res., 1993, 53, 3919–3924 Search PubMed.
- M. D. Reuber and C. W. Lee, Effect of age and sex on hepatic lesions in Buffalo strain rats ingesting diethylnitrosamine, J. Natl. Cancer Inst., 1968, 41, 1133–1140 Search PubMed.
- H. W. Rumsfeld Jr., W. L. Miller Jr. and C. A. Baumann, A sex difference in the development of liver tumors in rats fed 3′-methyl-4-dimethylaminoazobenzene or 4′-fluoro-4-dimethylaminoazobenzene, Cancer Res., 1951, 11, 814–819 Search PubMed.
- G. N. Wogan and P. M. Newberne, Dose-response characteristics of aflatoxin B1 carcinogenesis in the rat, Cancer Res., 1967, 27, 2370–2376 Search PubMed.
- H. Sidransky, B. P. Wagner and H. P. Morris, Sex difference in liver tumorgenesis in rats ingesting N-2-fluorenylacetamide, J. Natl. Cancer Inst., 1961, 26, 151–187 Search PubMed.
- F. De Matteis, C. Hollands, A. H. Gibbs, N. de Sa and M. Rizzardini, Inactivation of cytochrome P-450 and production of porphyrins caused in isolated hepatocytes by substituted dihydropyridines. Structural requirements for the loss of haem and alkylation of the pyrrole nitrogen atom, FEBS Lett., 1982, 145, 87–92 CrossRef PubMed.
- A. G. Smith and J. E. Francis, Chemically-induced formation of an inhibitor of hepatic uroporphyrinogen decarboxylase in inbred mice with iron overload, Biochem. J., 1987, 246, 221–226 CrossRef PubMed.
- A. G. Smith, J. E. Francis, S. J. Kay and J. B. Greig, Hepatic toxicity and uroporphyrinogen decarboxylase activity following a single dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin to mice, Biochem. Pharmacol., 1981, 30, 2825–2830 CrossRef PubMed.
- M.-T. Tsai, S.-Y. Huang and S.-Y. Cheng, Lead poisoning can be easily misdiagnosed as acute porphyria and nonspecific abdominal pain, Case Rep. Emerg. Med., 2017, 9050713 Search PubMed.
- S. Lindstedt, E. Holme, E. A. Lock, O. Hjalmarson and B. Strandvik, Treatment of hereditary tyrosinaemia type 1 by inhibition of 4-hydroxyphenylpyruvate dioxygenase, Lancet, 1992, 340, 813–817 CrossRef.
- A. G. Weinberg, C. E. Mize and H. G. Worthen, The occurrence of hepatoma in the chronic form of hereditary tyrosinemia, J. Pediatr., 1976, 88, 434–438 CrossRef PubMed.
- F. Angileri, V. Roy, G. Morrow, J. Y. Scoazec, N. Gadot, D. Grejuela and R. M. Tanguay, Molecular changes associated with chronic liver damage and neoplatic lesions in a murine model of hereditary tyrosinemia type1, Biochim. Biophys. Acta, 2015, 1852, 2603–2617 CrossRef PubMed.
- A. G. Smith, Porphyrias caused by chlorinated AH receptor ligands and associated mechanisms of liver injury and cancer, in The Porphyrin Handbook, Vol 14, Medical aspects of porphyrins, ed. K. I. Kadish, K. M. Smith & and R. D. Guilard, Academic Press, Amsterdam, 2003, ch. 92, pp. 169–210 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |