
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymer-based multiferroic nanocomposites via directed block copolymer self-assembly†
Ivan
Terzić
,
Niels L.
Meereboer
,
Harm Hendrik
Mellema
and
Katja
Loos
 *
*
Macromolecular Chemistry and New Polymeric Materials, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747AG Groningen, The Netherlands. E-mail: k.u.loos@rug.nl
First published on 21st December 2018
Abstract
The existence of ferroelectricity and ferromagnetism in multiferroic materials and their coupling enables the manipulation of the electric polarization with applied magnetic field and vice versa, opening many doors for the practical applications. However, the preparation of polymeric multiferroic nanocomposites is often accompanied with aggregation of magnetic particles inside the ferroelectric polymeric matrix. To overcome this issue, we developed a simple and straightforward method to obtain multiferroic nanocomposites with an exceptional and selective dispersion of magnetic nanoparticles, using self-assembly of poly(vinylidene fluoride) (PVDF)-based block copolymers. Magnetic cobalt ferrite nanoparticles modified with gallic acid are selectively incorporated within poly(2-vinylpyridine) (P2VP) domains of the lamellar block copolymer due to strong hydrogen bond formation between the ligand and the P2VP block. Using this approach, phase separation between the blocks is improved, which leads to an increase in the degree of crystallinity, whereas the selective dispersion of nanoparticles inside amorphous domains prevents changes in the crystalline phase of the ferroelectric block. The obtained nanocomposites demonstrate both ferroelectric and magnetic properties without large conductive losses at high electric field, making them good candidates for improved multiferroic devices.
Introduction
The concept of having at least two of the primary ferroic orders, that is, ferromagnetism, ferroelectricity or ferroelasticity, inside one material enables the use of multiferroics (MF) for numerous applications, such as sensors, transducers, actuators, data storage and energy harvesting devices.1–6 In particular, the coupling between ferroelectricity and ferromagnetism – the magnetoelectric (ME) effect – grants novel properties absent in each individual component, i.e. an electric polarization variation upon applying a magnetic field or inducing a magnetic response under an electric field.7,8 So far, two types of multiferroic materials have been investigated: single-phase and multiferroic nanocomposites.9–11 Single-phase multiferroics are relatively rare and exhibit weak ME responses at room temperature, which limits their potential application. The magnetoelectric response obtained in the MF composites is several orders of magnitude higher than for single-phase materials, which opens a door to a new generation of devices.12,13 Besides commonly used ceramic composites, polymer-based multiferroic materials, which combine piezoelectric poly(vinylidene fluoride) (PVDF) and inorganic magnetostrictive phases, have been attracting a growing research interest since they can be readily fabricated by cheap low temperature processing methods giving flexible materials with lower leakage currents and dielectric losses.4,14–18It has been established, both theoretically and experimentally, that the adhesion quality between the two phases and uniform filler dispersion are the determining factors for the creation of high quality multiferroic nanocomposites.13,15 However, preparing nanocomposites based on fluorinated polymers is a serious issue. The dense packing of fluorine atoms causes a low surface energy of PVDF, which results in strong demixing of this polymer with most inorganic fillers.19 The uneven distribution of the nano-objects inside the polymer matrix, as well as their aggregation, reduce the contact area between them and the polymer, causing a reduced ME coupling. Additionally, the aggregation of nano-objects is inevitably accompanied by increased conduction losses that lead to electric failure at low fields.20,21
Recently, many attempts have been made to produce PVDF-based nanocomposites with good dispersion of nano-objects, mostly focused on performing a surface modification on the nano-objects using, among others, silanes, phosphonic acid or PVDF polymer chains.22–24 However, little attention has been dedicated to the functionalization of the polymer matrix instead. So far, the incorporation of functional groups in the ferroelectric polymer that can strongly interact with the nano-object interface is exclusively achieved by the copolymerization of VDF with functional monomers or PVDF end group functionalization.20,25 Nevertheless, a small amount of comonomers can copolymerize with VDF, often yielding non-ferroelectric polymers as a result of an impaired crystallization or changed chain conformation.19 In contrary, chain end functionalization, even though without any significant impact on the crystallization process, often does not provide enough functionality to prevent nano-object aggregation.20
A very elegant method for introducing multiple functionalities into the structure of the ferroelectric polymer without impairing the crystallization is the preparation of PVDF-based block copolymers.26–28 Even though the presence of the amorphous block causes the reduction in the number of ferroelectric dipoles inside the material, the ability of block copolymers to self-assemble into different morphologies on the nanometer scale adds novel properties to the multiferroic nanocomposites. The self-assembly of block copolymers allows exact control over the nanocomposite morphology, local environment and polymer–particle interaction, which is difficult to achieve using pristine polymers.29–32 In addition, the selective confinement of nano-objects inside block copolymer nanodomains grants the possibility to achieve anisotropic properties that depend on the direction of the nanodomain alignment.33–35
The distribution of nano-objects inside block copolymers is a result of thermodynamic equilibrium between enthalpic and entropic contributions.36,37 Enthalpic contributions are related to the interaction between nano-objects and the functional block, whereas the selective incorporation of nano-objects inside block copolymers causes the reduction in entropy due to chain stretching and the loss in the translational motion of nano-objects.38–40 Therefore, attractive interactions, such as hydrogen bonding or ionic interactions, are crucial to overcome the entropic penalty and grant good and selective dispersion of nano-objects.41–43 Indeed, it has been demonstrated recently that strong hydrogen bonding between the surface of the nano-object and monomer units of the functional block enables high loading up to 40 wt% of nanoparticles without disrupting the phase separation of block copolymers.38,44
Herein, we report a straightforward route for the fabrication of multiferroic nanocomposites based on block copolymer self-assembly (Scheme 1). Selective dispersion of magnetic cobalt ferrite nanoparticles is achieved by forming strong hydrogen bonds between the ligands on the surface of the nanoparticles and the functional P2VP block of self-assembled poly(2-vinylpyridine)-b-poly(vinylidene fluoride-co-trifluorothylene)-b-poly(2-vinylpyridine) (P2VP-b-P(VDF-TrFE)-b-P2VP) triblock copolymers. The loading concentration of nanoparticles can reach 50 wt% in the targeted P2VP domain without drastically disrupting the self-assembled morphology. In this way, using block copolymer self-assembly together with strong hydrogen bond formation provides a new way to obtain multiferroic nanocomposites that show both ferroelectric and magnetic behavior without significant conduction losses at high electric fields.
 | ||
| Scheme 1 Schematic representation of the block copolymer self-assembly method for the preparation of multiferroic nanocomposites. The prepared nanocomposites possess both ferroelectric hysteresis of polarization (P) with changing the electric field (E) and superparamagnetic properties with zero coercive field in the magnetization (M)–magnetic field (H) curve. | ||
Experimental
Materials
1-Octadecene (90%, technical), FeCl3·6H2O (99+%, extra pure), CoCl2·6H2O (98–102%) and gallic acid (98%) were purchased from Acros Organics. Oleic acid (90%, technical) was purchased from Sigma Aldrich, whereas sodium oleate (97%, technical) was obtained from TCI Europe. All chemicals were used as received. The block copolymer was prepared following literature procedure.45 All solvents used for the film preparation and ligand exchange were analytical grade and used without further purification.Synthesis of (Co2+ Fe23+)-oleate precursor
In a mixture of 40 mL of water, 40 mL of ethanol and 80 mL of hexane, FeCl3·6H2O (4.32 mg, 16 mmol), CoCl2·6H2O (1.90 g, 8 mmol) and sodium oleate (19.48 g, 64 mmol) were added drop-wisely. The dark brow solution was stirred for 4 hours at 60 °C. The product was washed with water three times to remove the unreacted salt. Subsequently, the majority of solvent was removed using rotary evaporation, while the final traces were removed by drying under high vacuum (10−3 mbar) overnight.Synthesis of cobalt ferrite (CoFe2O4) nanoparticles
Precursor (2.5 g), oleic acid (0.25 g) and 1-octadecene (10 mL) were mixed in a triple neck flask equipped with a condenser. In order to remove final traces of water, the mixture was stirred at 80 °C under vacuum (10−3 mbar) for 1 h. Afterwards, nitrogen was slowly purged through the reaction mixture while it was heated to 310 °C at rate 1 °C min−1. The reaction mixture was maintained at 310 °C for 30 min and after cooling down to room temperature and washing three times with acetone, cobalt ferrite nanoparticles were obtained. The nanoparticles were dispersed in tetrahydrofuran and stored in the fridge.Ligand exchange and preparation of hydrophilic nanoparticles
A solution of gallic acid in tetrahydrofuran (500 mg in 10 mL) was added drop-wisely inside 10 mL of the solution of nanoparticles in tetrahydrofuran (c = 5 mg mL−1). To promote the ligand exchange, the obtained solution was kept in an ultrasonication bath overnight. The prepared nanoparticles were purified by precipitation with hexane and collected using centrifugation. The following procedure is repeated three times. The purified nanoparticles are readily dispersed in DMF at c = 5 mg mL−1 and kept in the fridge until further use.Preparation of polymer films
The block copolymer was dissolved in 4 mL DMF (10 mg mL−1) and the desired amount of nanoparticles was added. After passing through the 0.45 μm PTFE filter, the solution was cast in an aluminum pan (∅ 30 mm). The solvent was allowed to evaporate at 45 °C over two days. Subsequently, the film was heated to 170 °C during 5 min to induce the microphase separation. After cooling down and water lift-off, ca. 20 μm thick free-standing films were obtained. Any longer annealing of nanocomposite films in the melt results in the strong adhesion of films to the substrate and the pealing-off process is impossible to perform.Characterization
Small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS) measurements were carried out at the Dutch-Belgium Beamline (DUBBLE) station BM26B of the European Synchrotron Radiation Facility (ESRF) in Grenoble, France, particularly optimized for polymer investigation.46–48 The sample-to-detector distance was ca. 3.5 m for SAXS, ca. 28 cm for WAXS and an X-ray wavelength λ = 0.97 Å was used. SAXS images were recorded using a Pilatus 1 M detector while WAXS images were recorded using a Pilatus 100 kW detector, both with pixel size 172 × 172 μm. The scattering angle scale was calibrated using the known peak position from a standard silver behenate sample. The scattering intensity is reported as a function of the scattering vector q = 4π/λ(sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ) with 2θ being the scattering angle and λ the wavelength of the X-rays. Transmission electron microscopy (TEM) was performed on a Philips CM12 transmission electron microscope operating at an accelerating voltage of 120 kV. A piece of film was embedded in epoxy resin (Epofix, Electron Microscopy Sciences) and microtomed using a Leica Ultracut UCT-ultramicrotome in order to prepare ultrathin sections (ca. 80 nm). Enhanced contrast for nanocomposite samples was achieved using iodine staining of thin sections for 40 minutes. Nanoparticle samples were prepared by drop-casting diluted NP suspension onto carbon-supported copper grids. Fourier transform infrared (FTIR) spectra of cobalt ferrite nanoparticles and gallic acid were recorded using a Bruker Vertex 70 spectrophotometer in ATR mode with 32 scans at a nominal resolution of 4 cm−1. The D–E hysteresis measurements were performed using a state-of-the-art ferroelectric–piezoelectric tester aixACCT equipped with a Piezo Sample Holder Unit with a high voltage amplifier (0–10 kV). The AC electric field with a triangular wave form at frequency of 10 Hz was applied over polymer films immersed in silicon oil. The 100 nm thick gold electrodes (ca. 12.5 mm2) with 5 nm chromium adhesion layer were evaporated onto both sides. Magnetic hysteresis measurements were performed using a Quantum Design superconducting quantum interference device (SQUID) magnetic property measurement system (MPMS) at 300 K with a magnetic field up to 30 kOe.
θ) with 2θ being the scattering angle and λ the wavelength of the X-rays. Transmission electron microscopy (TEM) was performed on a Philips CM12 transmission electron microscope operating at an accelerating voltage of 120 kV. A piece of film was embedded in epoxy resin (Epofix, Electron Microscopy Sciences) and microtomed using a Leica Ultracut UCT-ultramicrotome in order to prepare ultrathin sections (ca. 80 nm). Enhanced contrast for nanocomposite samples was achieved using iodine staining of thin sections for 40 minutes. Nanoparticle samples were prepared by drop-casting diluted NP suspension onto carbon-supported copper grids. Fourier transform infrared (FTIR) spectra of cobalt ferrite nanoparticles and gallic acid were recorded using a Bruker Vertex 70 spectrophotometer in ATR mode with 32 scans at a nominal resolution of 4 cm−1. The D–E hysteresis measurements were performed using a state-of-the-art ferroelectric–piezoelectric tester aixACCT equipped with a Piezo Sample Holder Unit with a high voltage amplifier (0–10 kV). The AC electric field with a triangular wave form at frequency of 10 Hz was applied over polymer films immersed in silicon oil. The 100 nm thick gold electrodes (ca. 12.5 mm2) with 5 nm chromium adhesion layer were evaporated onto both sides. Magnetic hysteresis measurements were performed using a Quantum Design superconducting quantum interference device (SQUID) magnetic property measurement system (MPMS) at 300 K with a magnetic field up to 30 kOe.
Results and discussion
The formation of the polar crystalline phase with all-trans conformation of the P(VDF-TrFE) chains allows dipoles of the C–F bonds to flip in the direction of the applied electric field, while the high packing density prevents their misalignment after field removal, which causes hysteretic ferroelectric behavior of this material.49 The addition of insulating chains to the ferroelectric polymer structure, in the form of graft or block copolymers, has been proven to induce changes in the switching nature and the shape of the hysteresis loops of P(VDF-TrFE).50 Recently, we have demonstrated that the incorporation of the polar P2VP at chain ends of P(VDF-TrFE) and the formation of the lamellar morphology via the self-assembly of the block copolymer governs the preservation of the ferroelectric hysteresis loop with switching characteristics similar to the neat ferroelectric polymer.45 The additional benefit of choosing P2VP as an insulating block is related to P2VP's affinity to form hydrogen bonds, which is highly attractive for the inclusion of nanoparticles inside block copolymer matrices.41,51,52 The P2VP-b-P(VDF-TrFE)-b-P2VP block copolymer is synthesized following our recent procedure using a copper catalyzed azide/alkyne cycloaddition reaction of the alkyne terminated P2VP and the azide terminated (PVDF-TrFE).45 The low dispersity P2VP is prepared using reversible addition–fragmentation chain transfer (RAFT) polymerization, whereas free-radical copolymerization of the VDF and TrFE with chlorine-functionalized benzoyl peroxide initiators and the subsequent end group transformation are employed for the synthesis of the telechelic azide-terminated P(VDF-TrFE).53 Several block copolymers with different molecular weight and ratio between the blocks are prepared. Throughout this study, we focused on one block copolymer with Mn = 32.8 kg mol−1, dispersity index Đ = 1.80 and 30 wt% of P2VP inside the block copolymer. These molecular characteristics drive the self-assembly of the block copolymer into the lamellar morphology with P2VP domains slightly bigger in size than the diameter of nanoparticles, necessary for their unhindered selective dispersion inside P2VP domains.Cobalt ferrite (CFO) nanoparticles are prepared by thermal decomposition of an iron–cobalt oleate precursor in a high boiling-point solvent according to literature procedure.54 This method leads to highly monodisperse nanoparticles with sizes that are easily tuned by the adjustment of the inert gas flow through the reaction mixture.55 These nanoparticles are mainly chosen due to their magnetostriction value (λ = 220 ppm),56 which is higher than for other ferrites. The average core diameter of the prepared nanoparticles used for the nanocomposite preparation is 6.6 ± 0.4 nm (Fig. 1a). The surface of the as-synthesized nanoparticles is coated with a non-polar oleic acid layer inadequate for forming strong interactions with the P2VP, which is crucial to achieve high loading of the nanoparticles, while avoiding their aggregation. To enable the formation of the hydrogen bonds with the block copolymer, the long alkyl chains on the surface of nanoparticles are replaced with gallic acid.57 Gallic acid is particularly selected since it contains a carboxylic group with an affinity towards the nanoparticle surface, as well as three phenolic hydroxyl groups able to form strong hydrogen bonds with the polymer matrix, specifically with the P2VP side chains. The successful ligand exchange is confirmed by FTIR spectra (Fig. 1b) in which the shift of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration signal is clearly observed, suggesting that carboxylic groups are mainly employed in the bond formation with the nanoparticle core. Additionally, after the ligand exchange a wide peak at 3100–3400 cm−1 characteristic for O–H stretching from hydroxyl groups at the surface of nanoparticles is clearly demonstrated. The nanoparticles modified by gallic acid showed excellent dispersibility inside dimethylformamide (DMF) during long-time observation (1 year), since DMF forms hydrogen bonds with ligand molecules on the surface. It is worth noticing that the ligand exchange does not proceed quantitatively and some oleic acid molecules are present on the surface after ligand exchange. However, the presence of this non-polar ligands demonstrated no negative effect on the stability of nanoparticles and their dispersion in block copolymers.
O stretching vibration signal is clearly observed, suggesting that carboxylic groups are mainly employed in the bond formation with the nanoparticle core. Additionally, after the ligand exchange a wide peak at 3100–3400 cm−1 characteristic for O–H stretching from hydroxyl groups at the surface of nanoparticles is clearly demonstrated. The nanoparticles modified by gallic acid showed excellent dispersibility inside dimethylformamide (DMF) during long-time observation (1 year), since DMF forms hydrogen bonds with ligand molecules on the surface. It is worth noticing that the ligand exchange does not proceed quantitatively and some oleic acid molecules are present on the surface after ligand exchange. However, the presence of this non-polar ligands demonstrated no negative effect on the stability of nanoparticles and their dispersion in block copolymers.
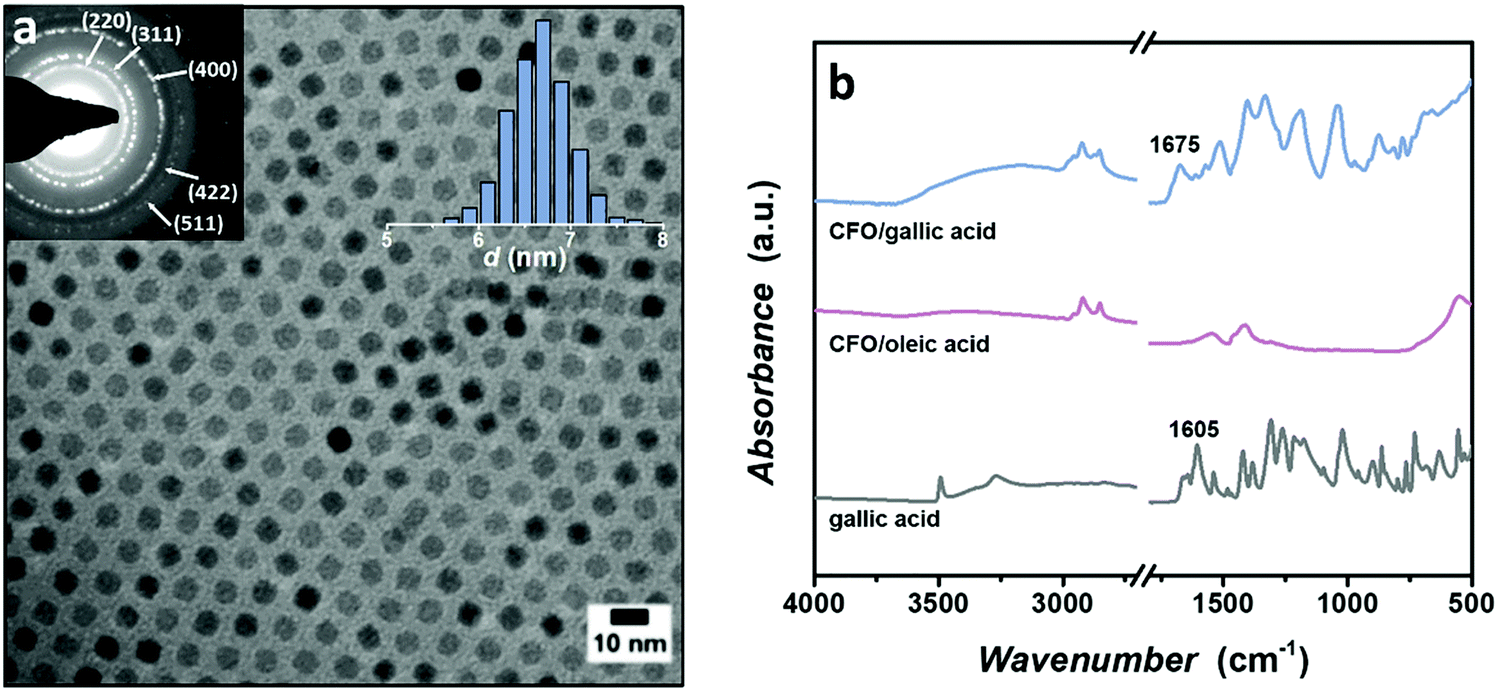 | ||
| Fig. 1 (a) TEM micrograph of the oleic acid capped cobalt ferrite nanoparticles with size distribution. The average diameter is 6.6 ± 0.4 nm, obtained using ImageJ image analysis. The selected area electron diffraction pattern corresponds to the spinel structure of cobalt ferrite. (b) FTIR spectra of gallic acid, nanoparticles covered with oleic acid and nanoparticles covered with gallic acid showing successful ligand exchange and the bonding between the surface of the nanoparticle and gallic acid. | ||
Small-angle X-ray scattering (SAXS) and transmission electron microscopy (TEM) are used to characterize the morphology and the location of nanoparticles inside the block copolymer domains.46,47 The block copolymer film is prepared in an aluminum petri dish by solvent casting 1 wt% polymer solution from DMF at 45 °C during 2 days and its subsequent thermal annealing at 170 °C above the melting point of P(VDF-TrFE) to obtain the equilibrium structure. After this, the polymer film is cooled down to induce crystallization of the ferroelectric block. For the nanocomposite films, the DMF dispersion of nanoparticles is mixed with the block copolymer solution in the desired ratio and the same processing conditions are applied; the concentration of nanoparticles is given as the weight percentage of the P2VP block based on the mass of nanoparticles with ligands. As depicted in Fig. 2a, the SAXS profile of the block copolymer displays three scattering peaks in ratio 1q*:2q*:3q*, revealing the lamellar morphology with the lamellar spacing dL = 2π/q* = 34 nm. The lamellar nanostructure is a result of the P(VDF-TrFE) crystallization confinement inside lamellar domains resulting from the block copolymer self-assembly, which is confirmed by the similarity in shape of the SAXS signal before and after crystallization. The structure is furthermore proved by the non-stained TEM image of the microtomed block copolymer sample (Fig. 3a) that shows a well-ordered lamellar morphology in which dark layers correspond to the P(VDF-TrFE) crystalline phase, while the amorphous P2VP phase appears light. In contrast to the block copolymer, the ordering of the nanocomposites into the lamellar structure has become worse, as can be observed by broadening of the first order and disappearance of the larger order scattering peaks after nanoparticle addition (Fig. 2b). The incorporation of nanoparticles inside the block copolymer system reduces the mobility of polymer chains in the melt. Thus, short times necessary to obtain well-ordered lamellar morphology inside pristine block copolymer are insufficient to achieve high ordering of nanocomposites. However, still well-ordered lamellar morphologies are observed with nanoparticle arrays up to concentrations, c = 30 wt% compared to the P2VP block (Fig. 3b–d). At concentration c = 50 wt% the block copolymer nanocomposite still self-assembles into the lamellar structure, although with a significantly decreased long-range ordering. The addition of such a large number of nanoparticles results in smaller isolated ordered lamellar domains with regions in which the migration of nanoparticles inside P(VDF-TrFE) domains can be observed.
 | ||
| Fig. 2 (a) SAXS profile for a block copolymer collected at 170 °C and at room temperature showing the formation of the lamellar structure in the melt and its preservation after the crystallization of the P(VDF-TrFE). (b) SAXS profiles of block copolymer/CFO nanocomposites at different loading of nanoparticles. | ||
 | ||
| Fig. 3 TEM images of (a) pristine block copolymer and nanocomposites with (b) 10 wt%, (c) 20 wt%, (d) 30 wt%, (e) 50 wt% of nanoparticles compared to the total weight of P2VP domains demonstrating significantly improved and selective dispersion of nanoparticles due to the hydrogen bond formation. (f) Dispersion of gallic acid coated CFO nanoparticles inside the pure P(VDF-TrFE) showing the macrophase separation of nanoparticles from the polymer. | ||
Fig. 3b–e demonstrate the distribution of gallic acid coated CFO nanoparticles inside the block copolymer with increased loading of nanoparticles. An exclusive arrangement of nanoparticles can be observed inside P2VP domains that appears dark in TEM images due to iodine staining. Such a specific control of nanoparticles distribution inside P2VP domains is evidently the consequence of the strong hydrogen bond formation between the hydroxyl groups on the surface of nanoparticles and the P2VP units. Another beneficial factor that affects the good selective distribution of the nanoparticles inside this specific block copolymer is related to the crystallization of the P(VDF-TrFE), that can expel nanoparticles from the crystalline domains.58 Note that the nanocomposite of pristine P(VDF-TrFE) and gallic acid coated nanoparticles exhibits macrophase separation with micrometer sized aggregates of nanocrystals in the polymer (Fig. 3f). Even though the hydrogen bond formation between hydroxyl groups and VDF segments has been already demonstrated in literature,59 the intensity of these bonds is apparently too weak to induce satisfactory dispersion of cobalt ferrite nanoparticles.
The mixing of the block copolymer and CFO nanoparticles results in an increase of the lamellar domain spacing, as indicated by a shift of the scattering maximum to lower q-values (Fig. 2b). The size of the lamellar spacing increases from 34 nm to 42 nm after the incorporation of 30 wt% of nanoparticles, due to the selective swelling of the amorphous P2VP domains. In order to further demonstrate the strength of this method for the spatial distribution and dispersion of nanoparticles, bigger nanoparticles with core diameter d = 12.5 nm and d/L = 0.85 are mixed with the block copolymer at concentration c = 30 wt% (Fig. S3, ESI†). No significant difference in the dispersion and location of nanoparticles is observed (Fig. S3, ESI†). Small regions of the aggregated nanoparticles are mainly a consequence of the insufficient stability of bigger nanoparticles in DMF. It can be observed that the stability of these nanoparticles during film casting is fairly reduced compared to the smaller nanoparticles. The addition of a different ligand with a larger number of functional groups or, alternatively, covering the surface with a functional polymer layer can be a potential way to prevent the undesired particle aggregation and obtain high quality nanocomposites with even bigger nanoparticles.44 However, this specific block copolymer cannot be used for the dispersion of nanoparticles bigger than its domain size, which can be easily solved by using a block copolymer with a higher molecular weight.
The crystallization behavior of nanocomposites and the influence of the nanoparticle addition on the chain conformation of the ferroelectric block are investigated using differential scanning calorimetry (DSC) and wide-angle X-ray scattering (WAXS). Fig. 4a displays the crystallization exotherms of the pristine block copolymer and the corresponding nanocomposite with 30 wt% of nanoparticles. No difference in shape of the crystallization exotherms for nanocomposites with different loading of nanoparticles is observed. Both DSC curves show two exotherms, where the signal at higher temperature corresponds to the crystallization into a paraelectric phase which is followed by a paraelectric-to-ferroelectric Curie transition at 51 °C.49 Contrary to the neat block copolymer, for which a wide crystallization peak is observed, a much sharper signal at a slightly higher temperature is characteristic for the nanocomposites, mainly due to a stronger domain separation after the addition of nanoparticles. An increase in the segregation strength between the blocks or even the induction of the microphase separation of otherwise disordered block copolymers has been already demonstrated after the addition of nanoparticles or other fillers that can form selective hydrogen bonds with one of the blocks.58,61 The same effect can explain the rise in P(VDF-TrFE) crystallinity after selective dispersion of nanoparticles up to 30 wt% loading, demonstrating a profound effect of nanoparticle addition on the crystallization of the crystalline block. As observed in Fig. 3e, a slight aggregation of nanoparticles and migration to the crystalline layers take place at loadings as high as 50 wt%, which results in the reduction of the degree of crystallinity (Table 1).
 | ||
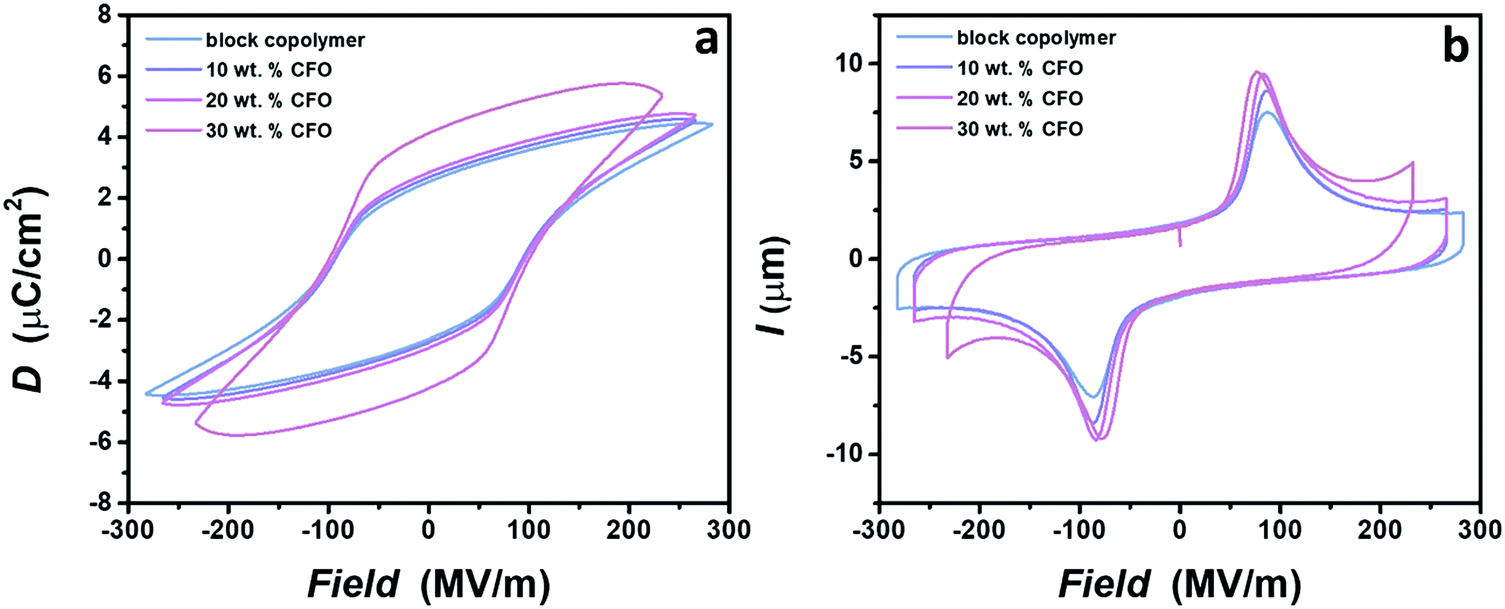
| Fig. 4 (a) D–E loops and (b) I–E curves for a block copolymer and nanocomposites. No significant difference in the shape of the hysteresis is observed for nanocomposites with loading up to 20 wt%. Drastic increase in the polarization for the nanocomposite with 30 wt% is a consequence of increased conductive losses. However, nanocomposite still operates at high electric fields. It is shown that the coercive field increases with the applied electric field. Therefore, the reduced coercive field for nanocomposites with 30 wt% of CFO is a consequence of the lower electric field used for dipole switching. | ||
| Sample name | ΔHCa (J g−1) | X C (%) | Crystalline phasec |
|---|---|---|---|
| a Determined using DSC. b Degree of crystallinity calculated using the following equation: XC = (ΔHC)/(fP(VDF-TrFE)·ΔH100) × 100%. ΔHC was determined based on DSC thermograms. ΔH100 = 42 J g−1 for crystallization in the paraelectric phase.60 c Determined using WAXS. | |||
| Block copolymer (BCP) | 11.5 | 39.1 | LTFE |
| BCP/10 wt% CFO | 12.5 | 44.4 | LTFE |
| BCP/20 wt% CFO | 11.9 | 44.2 | LTFE |
| BCP/30 wt% CFO | 11.1 | 43.1 | LTFE |
| BCP/50 wt% CFO | 7.7 | 33.3 | LTFE + HTPE |
The addition of different nano-objects to the ferroelectric polymer has been proven to generate changes in its crystalline phases. Therefore, the crystalline nature of nanocomposites is also examined using WAXS (Fig. 4b).48 The incorporation of nanoparticles inside P2VP layers has no effect on the crystalline phase and chain conformation of the P(VDF-TrFE) up to 30 wt% of nanoparticles, as expected for a system in which phase separation between nanoparticles and crystalline layers occurs. For a block copolymer and nanocomposites up to 30 wt% loading, only one scattering peak at q = 14.0 nm−1 is observed, corresponding to the low temperature ferroelectric phase (LTFE) with all-trans conformation of polymer chains.62 A further increase in the nanoparticle concentration leads to the formation of an additional high temperature paraelectric phase with trans–gauche conformation.62 Since at high loadings aggregation and migration of nanoparticles in crystalline domains occur, the functional groups on the nanoparticles’ surface can induce defects and conformational changes of the ferroelectric polymer chains. Similar conformational changes are noticed upon the addition of polar fillers-zeolites, clays or polar miscible polymer chains.63–65 Therefore, selective dispersion of nanoparticles inside block copolymers is a more appealing method to preserve the desired crystalline phase necessary for ferroelectric dipole switching.
Multiferroic nanocomposites possess two ferroic orders inside one material: ferroelectricity and ferromagnetism. Thus, it is highly important to examine the ferroelectricity of nanocomposites and the effect of the nanoparticle incorporation on the dipole switching behavior. The ferroelectric response is determined using displacement–electric field (D–E) and current–electric field (I–E) loop measurements with the bipolar triangular waveform applied at the frequency of 10 Hz, slightly below breakdown electric fields. All samples show ferroelectric behavior, demonstrated by a peak on the I–E curve and the rectangular shape of the hysteresis loop. Note that the ferroelectric response is not shown for the nanocomposite with 50 wt% of nanoparticles, since such a high loading of ferrite nanoparticles leads to brittle films with a low breakdown strength caused by the formation of a continuous conductive network inside material, as demonstrated in Fig. 3e. Fig. 4a shows that no substantial differences are detected in the shape of the hysteresis loop for the neat block copolymer and nanocomposites with 10 and 20 wt% of nanoparticles. Only a slight increase of the maximum polarization, Pmax, values is observed with the addition of nanoparticles, probably caused by the rise in the crystallinity and the dielectric constant after the incorporation of cobalt ferrite nanoparticles.59 An increase in the polarization is followed by the increase in the intensity of the current peak on the I–E curve (Fig. 4b). In contrast to these samples, a strong increase of the Pmax is demonstrated for the nanocomposite with 30 wt% loading. Two effects can explain this difference in behavior. As displayed in Fig. 3d, the concentration of nanoparticles tends to a percolation threshold, which leads to increased conductive losses and therefore higher values of polarization.20,66 Indeed, higher values of current are observed at a high field for this nanocomposite sample compared to the others (Fig. 4b). However, even with the increased losses, this sample reached similar electric fields as the neat block copolymer. Additionally, the strong jump of the dielectric constant near the percolation threshold, previously demonstrated in various nanocomposites, can be a probable cause for an increase in polarization values.67,68 Despite different values of the Pmax, no difference in coercive field is observed between samples, which is of great significance, since the application of nanocomposites is still related to the lower electric fields that are easier to achieve and safer to operate at.
Fig. 5 shows the magnetization loops of all samples with a different cobalt ferrite content. The hysteresis loop of the neat block copolymer reveals the expected diamagnetic behavior, while the incorporation of cobalt ferrite nanoparticles induces changes in the magnetic response of the nanocomposites. All samples show a strong interaction with the magnetic field, in which saturation magnetization increases gradually on the addition of more nanoparticles. However, no coercive field is observed regardless of the concentration of nanoparticles, because the size of cobalt ferrite nanoparticles is below the critical size (dc = 10 nm), under which the superparamagnetic behavior is expected.69 Each nanoparticle consists of a single magnetic domain and when placed in the magnetic field, it develops a strong interaction with the field. Since only one domain is contained in one particle, no cooperative interaction between domains characterizes the prepared nanocomposites. Accordingly, no net magnetization is preserved after the removal of the magnetic field.70 Employing a magnetic component with no hysteresis loop into multiferroic composites attracts much research interest, especially in the magnetic sensor application, since it generates devices with low noise and high sensitivity.16 The hysteresis loop can be achieved undoubtedly on demand by changing the size, aspect ratio or chemical structure of nanoparticles. However, this is out of the scope of this paper.
 | ||
| Fig. 5 Magnetization versus applied magnetic field for the block copolymer and nanocomposites at 300 K with a maximum applied field of 30 kOe. No hysteretic behavior is demonstrated for any sample. | ||
Conclusions
The preparation of polymer multiferroic nanocomposites with exceptional dispersion of magnetic nano-objects inside the ferroelectric polymer matrix is highly desirable for various high-performance applications, from sensors and transducers to data storage and energy harvesting devices. However, this has demonstrated to be a challenging task, mostly due to strong immiscibility of PVDF with various inorganic materials. Here, we present a direct and simple method for the fabrication of multiferroic nanocomposites with homogeneous dispersion of nanoparticles using a block copolymer self-assembly approach. The strong hydrogen bond formation between gallic acid coated cobalt ferrite nanoparticles and functional P2VP block of P2VP-b-P(VDF-TrFE)-b-P2VP block copolymer grants selective dispersion of magnetic nanoparticles inside P2VP layers of block copolymer lamellar domains. The selective incorporation of nanoparticles inside non-ferroelectric block is confirmed to be beneficial for the preservation of the ferroelectric crystalline phase and a higher degree of crystallinity of ferroelectric block. Using this straightforward method, multiferroic nanocomposites, enjoying both unaffected ferroelectric behavior and strong response to magnetic field with zero coercive field, are easily accomplished. They can be potentially employed in magnetic sensor devices with improved sensitivity and signal-to-noise ratio after further optimization (i.e., alignment of lamellar domains, other block copolymer morphologies, content of the amorphous block, size of the block copolymer).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Netherlands Organization for Scientific Research (NWO) via a VICI innovational research grant. The authors are very grateful to Prof. Beatriz Noheda for the valuable discussion regarding the ferroelectric measurements and to Jacob Baas for the assistance with magnetic measurements. Beam time on the Dutch-Belgian Beamline (DUBBLE) of ESRF (Grenoble, France) has kindly been made available by NWO, and we would like to thank Daniel Hermida-Merino for his experimental assistance.Notes and references
- W. Eerenstein, N. D. Mathur and J. F. Scott, Nature, 2006, 442, 759–765 CrossRef CAS PubMed.
- M. Fiebig, T. Lottermoser, D. Meier and M. Trassin, Nat. Rev. Mater., 2016, 1, 16046 CrossRef CAS.
- Y. Chen, S. M. Gillette, T. Fitchorov, L. Jiang, H. Hao, J. Li, X. Gao, A. Geiler, C. Vittoria and V. G. Harris, Appl. Phys. Lett., 2011, 99, 042505 CrossRef.
- P. Martins and S. Lanceros-Méndez, Adv. Funct. Mater., 2013, 23, 3371–3385 CrossRef CAS.
- T.-D. Onuta, Y. Wang, C. J. Long and I. Takeuchi, Appl. Phys. Lett., 2011, 99, 203506 CrossRef.
- N. A. Spaldin and M. Fiebig, Science, 2005, 309, 391–392 CrossRef CAS PubMed.
- M. Fiebig, J. Phys. D: Appl. Phys., 2005, 38, R123 CrossRef CAS.
- C.-W. Nan, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 6082–6088 CrossRef CAS.
- W. Prellier, M. P. Singh and P. Murugavel, J. Phys.: Condens. Matter, 2005, 17, R803 CrossRef CAS.
- X. Gao, B. J. Rodriguez, L. Liu, B. Birajdar, D. Pantel, M. Ziese, M. Alexe and D. Hesse, ACS Nano, 2010, 4, 1099–1107 CrossRef CAS PubMed.
- R. Comes, H. Liu, M. Khokhlov, R. Kasica, J. Lu and S. A. Wolf, Nano Lett., 2012, 12, 2367–2373 CrossRef CAS PubMed.
- L. Van Lich, T. Shimada, K. Miyata, K. Nagano, J. Wang and T. Kitamura, Appl. Phys. Lett., 2015, 107, 232904 CrossRef.
- J. Jin, S.-G. Lu, C. Chanthad, Q. Zhang, M. A. Haque and Q. Wang, Adv. Mater., 2011, 23, 3853–3858 CAS.
- T. Prabhakaran and J. Hemalatha, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 2418–2422 CrossRef CAS.
- M. Silva, S. Reis, C. S. Lehmann, P. Martins, S. Lanceros-Mendez, A. Lasheras, J. Gutiérrez and J. M. Barandiarán, ACS Appl. Mater. Interfaces, 2013, 5, 10912–10919 CrossRef CAS PubMed.
- P. Martins, Y. V. Kolen’ko, J. Rivas and S. Lanceros-Mendez, ACS Appl. Mater. Interfaces, 2015, 7, 15017–15022 CrossRef CAS PubMed.
- V. S. D. Voet, M. Tichelaar, S. Tanase, M. C. Mittelmeijer-Hazeleger, G. ten Brinke and K. Loos, Nanoscale, 2013, 5, 184–192 RSC.
- V. S. D. Voet, D. Hermida-Merino, G. ten Brinke and K. Loos, RSC Adv., 2013, 3, 7938–7946 RSC.
- B. Ameduri, Chem. Rev., 2009, 109, 6632–6686 CrossRef CAS PubMed.
- J. Li, P. Khanchaitit, K. Han and Q. Wang, Chem. Mater., 2010, 22, 5350–5357 CrossRef CAS.
- T. Zhou, J.-W. Zha, R.-Y. Cui, B.-H. Fan, J.-K. Yuan and Z.-M. Dang, ACS Appl. Mater. Interfaces, 2011, 3, 2184–2188 CrossRef CAS PubMed.
- K. Prabakaran, S. Mohanty and S. K. Nayak, J. Mater. Sci.: Mater. Electron., 2014, 25, 4590–4602 CrossRef CAS.
- P. Kim, S. C. Jones, P. J. Hotchkiss, J. N. Haddock, B. Kippelen, S. R. Marder and J. W. Perry, Adv. Mater., 2007, 19, 1001–1005 CrossRef CAS.
- R. Vukićević, I. Vukovic, H. Stoyanov, A. Korwitz, D. Pospiech, G. Kofod, K. Loos, G. ten Brinke and S. Beuermann, Polym. Chem., 2012, 3, 2261–2265 RSC.
- S. Banerjee, M. Wehbi, A. Manseri, A. Mehdi, A. Alaaeddine, A. Hachem and B. Ameduri, ACS Appl. Mater. Interfaces, 2017, 9, 6433–6443 CrossRef CAS PubMed.
- V. S. D. Voet, G. O. R. A. van Ekenstein, N. L. Meereboer, A. H. Hofman, G. ten Brinke and K. Loos, Polym. Chem., 2014, 5, 2219–2230 RSC.
- V. S. D. Voet, G. ten Brinke and K. Loos, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2861–2877 CrossRef CAS.
- V. S. D. Voet, K. Kumar, G. ten Brinke and K. Loos, Macromol. Rapid Commun., 2015, 36, 1756–1760 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- G. Kortaberria and A. Tercjak, Block Copolymer Nanocomposites, Pan Stanford, 2016 Search PubMed.
- J. Kao, K. Thorkelsson, P. Bai, B. J. Rancatore and T. Xu, Chem. Soc. Rev., 2013, 42, 2654–2678 RSC.
- M. R. Bockstaller, R. A. Mickiewicz and E. L. Thomas, Adv. Mater., 2005, 17, 1331–1349 CrossRef CAS.
- Y. Rokhlenko, M. Gopinadhan, C. O. Osuji, K. Zhang, C. S. O’Hern, S. R. Larson, P. Gopalan, P. W. Majewski and K. G. Yager, Phys. Rev. Lett., 2015, 115, 258302 CrossRef PubMed.
- P. W. Majewski, M. Gopinadhan and C. O. Osuji, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 2–8 CrossRef CAS.
- M. J. Park and N. P. Balsara, Macromolecules, 2010, 43, 292–298 CrossRef CAS.
- V. Pryamitsyn and V. Ganesan, Macromolecules, 2006, 39, 8499–8510 CrossRef CAS.
- H. Kang, F. A. Detcheverry, A. N. Mangham, M. P. Stoykovich, K. C. Daoulas, R. J. Hamers, M. Müller, J. J. de Pablo and P. F. Nealey, Phys. Rev. Lett., 2008, 100, 148303 CrossRef PubMed.
- L. Yao, Y. Lin and J. J. Watkins, Macromolecules, 2014, 47, 1844–1849 CrossRef CAS.
- M. E. Mackay, A. Tuteja, P. M. Duxbury, C. J. Hawker, B. V. Horn, Z. Guan, G. Chen and R. S. Krishnan, Science, 2006, 311, 1740–1743 CrossRef CAS PubMed.
- P. Rittigstein, R. D. Priestley, L. J. Broadbelt and J. M. Torkelson, Nat. Mater., 2007, 6, 278–282 CrossRef CAS PubMed.
- Y. Gai, Y. Lin, D.-P. Song, B. M. Yavitt and J. J. Watkins, Macromolecules, 2016, 49, 3352–3360 CrossRef CAS.
- S. C. Warren, L. C. Messina, L. S. Slaughter, M. Kamperman, Q. Zhou, S. M. Gruner, F. J. DiSalvo and U. Wiesner, Science, 2008, 320, 1748–1752 CrossRef CAS PubMed.
- M. Golkaram, C. Fodor, E. van Ruymbeke and K. Loos, Macromolecules, 2018, 51, 4910–4916 CrossRef CAS PubMed.
- D.-P. Song, Y. Lin, Y. Gai, N. S. Colella, C. Li, X.-H. Liu, S. Gido and J. J. Watkins, J. Am. Chem. Soc., 2015, 137, 3771–3774 CrossRef CAS PubMed.
- I. Terzic, N. L. Meereboer, M. Acuautla, G. Portale and K. Loos, Nat. Commun., 2019 Search PubMed , peer review.
- W. Bras, I. P. Dolbnya, D. Detollenaere, R. van Tol, M. Malfois, G. N. Greaves, A. J. Ryan and E. Heeley, J. Appl. Crystallogr., 2003, 36, 791–794 CrossRef CAS.
- M. Borsboom, W. Bras, I. Cerjak, D. Detollenaere, D. Glastra van Loon, P. Goedtkindt, M. Konijnenburg, P. Lassing, Y. K. Levine, B. Munneke, M. Oversluizen, R. van Tol and E. Vlieg, J. Synchrotron Radiat., 1998, 5, 518–520 CrossRef CAS PubMed.
- G. Portale, D. Cavallo, G. C. Alfonso, D. Hermida-Merino, M. van Drongelen, L. Balzano, G. W. M. Peters, J. G. P. Goossens and W. Bras, J. Appl. Crystallogr., 2013, 46, 1681–1689 CrossRef CAS.
- T. Soulestin, V. Ladmiral, F. D. Dos Santos and B. Améduri, Prog. Polym. Sci., 2017, 72, 16–60 CrossRef CAS.
- F. Guan, L. Yang, J. Wang, B. Guan, K. Han, Q. Wang and L. Zhu, Adv. Funct. Mater., 2011, 21, 3176–3188 CrossRef CAS.
- S. Bondzic, J. de Wit, E. Polushkin, A. J. Schouten, G. ten Brinke, J. Ruokolainen, O. Ikkala, I. Dolbnya and W. Bras, Macromolecules, 2004, 37, 9517–9524 CrossRef CAS.
- A. H. Hofman, I. Terzic, M. C. A. Stuart, G. ten Brinke and K. Loos, ACS Macro Lett., 2018, 1168–1173 CrossRef CAS PubMed.
- I. Terzic, N. L. Meereboer and K. Loos, Polym. Chem., 2018, 9, 3714–3720 RSC.
- N. Bao, L. Shen, W. An, P. Padhan, C. Heath Turner and A. Gupta, Chem. Mater., 2009, 21, 3458–3468 CrossRef CAS.
- J. H. Jung, S. Kim, H. Kim, J. Park and J. H. Oh, Small, 2015, 11, 4976–4984 CrossRef CAS PubMed.
- D. Fritsch and C. Ederer, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 104117 CrossRef.
- X. Wang, R. D. Tilley and J. J. Watkins, Langmuir, 2014, 30, 1514–1521 CrossRef CAS PubMed.
- Y. Lin, X. Wang, G. Qian and J. J. Watkins, Chem. Mater., 2014, 26, 2128–2137 CrossRef CAS.
- J. Li, J. Claude, L. E. Norena-Franco, S. I. Seok and Q. Wang, Chem. Mater., 2008, 20, 6304–6306 CrossRef CAS.
- R. J. Klein, J. Runt and Q. M. Zhang, Macromolecules, 2003, 36, 7220–7226 CrossRef CAS.
- Y. Lin, V. K. Daga, E. R. Anderson, S. P. Gido and J. J. Watkins, J. Am. Chem. Soc., 2011, 133, 6513–6516 CrossRef CAS PubMed.
- Y. Li, T. Soulestin, V. Ladmiral, B. Ameduri, T. Lannuzel, F. D. D. Santos, Z.-M. Li, G.-J. Zhong and L. Zhu, Macromolecules, 2017, 50, 7646–7656 CrossRef CAS.
- A. C. Lopes, C. Caparros, S. Ferdov and S. Lanceros-Mendez, J. Mater. Sci., 2013, 48, 2199–2206 CrossRef CAS.
- A. C. Lopes, C. M. Costa, C. J. Tavares, I. C. Neves and S. Lanceros-Mendez, J. Phys. Chem. C, 2011, 115, 18076–18082 CrossRef CAS.
- N. L. Meereboer, I. Terzić, S. Saidi, D. Hermida Merino and K. Loos, ACS Macro Lett., 2018, 863–867 CrossRef CAS PubMed.
- L. Xie, X. Huang, K. Yang, S. Li and P. Jiang, J. Mater. Chem. A, 2014, 2, 5244–5251 RSC.
- S. Tu, Q. Jiang, X. Zhang and H. N. Alshareef, ACS Nano, 2018, 12, 3369–3377 CrossRef CAS PubMed.
- D. Wilkinson, J. S. Langer and P. N. Sen, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 1081–1087 CrossRef.
- C. P. Bean and J. D. Livingston, J. Appl. Phys., 1959, 30, S120–S129 CrossRef.
- A. Akbarzadeh, M. Samiei and S. Davaran, Nanoscale Res. Lett., 2012, 7, 144 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tc05017a |
| This journal is © The Royal Society of Chemistry 2019 |