DOI:
10.1039/C8TC04967J
(Paper)
J. Mater. Chem. C, 2018,
6, 13293-13304
Quaternary phosphonium-based (TPQPCl)-ionomer/graphite nanoplatelets composite chemically modified electrodes: a novel platform for sensing applications†
Received
1st October 2018
, Accepted 8th November 2018
First published on 3rd December 2018
Abstract
Ionomers have attracted considerable interest in electroanalysis due to the possibility of fabricating electrode coatings capable of preconcentrating sub-micromolar concentrations of cations or anions of analytical relevance. In this work, we describe the electroanalytical performances of an ionomer (TPQPCl)/graphite nanoplatelets composite material towards the development of an amperometric sensor for detection of ascorbic acid. Graphite nanoplatelets at different concentrations were dispersed in ethanolic solutions containing TPQPCl. The as-prepared TPQPCl/graphite nanoplatelet-coated electrodes were characterised using Fe(CN)64−/3− as an anionic redox probe. The results evidence the good preconcentration capability of the positively charged TPQPCl towards the incorporation of negatively charged species. By tuning the ionomer/graphite nanoplatelets ratio, it is possible to detect simultaneously ascorbic acid even in the presence of dopamine as an interference species. The TPQPCl/graphite nanoplatelet-coated electrodes were able to detect ascorbic acid in the linear range of 5–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μM with a limit of detection calculated as 4.8 μM using linear sweep voltammetry. Finally, the TPQPCl/graphite nanoplatelet-coated electrodes were tested towards detection of ascorbic acid in vitamin C tablets and orange juice without any sample pretreatment.
000 μM with a limit of detection calculated as 4.8 μM using linear sweep voltammetry. Finally, the TPQPCl/graphite nanoplatelet-coated electrodes were tested towards detection of ascorbic acid in vitamin C tablets and orange juice without any sample pretreatment.
1. Introduction
There is still considerable interest in the development of analytical devices able to detect chemical species with high sensitivity and selectivity. In this respect, electrochemical sensors are very appealing due to their ease of fabrication, fast response time and affordable cost,1,2 and have a broad range of applications spanning from detection of analytes for environmental monitoring,3,4 to food, and healthcare.5 Among the plethora of materials utilised as electrode surfaces for amperometric detection, carbon, in its different allotrope forms, is undoubtedly the most popular material. Typical examples include glassy carbon,6 carbon paste,7 and carbon nanotubes,8 among the most widely utilised electrodes. However, the discovery of graphene in 2004 by Novoselov and Geim9 has extended further the list of carbon allotropes suitable for electroanalytical applications, as evidenced by the huge amount of literature in this field in the last decade.10,11 Graphene is defined as a two-dimensional single layer of carbon atoms arranged in a honeycomb-like (hexagonal) lattice structure. The sp2 hybridisation of carbon bonds means that the in-plane σC–C bond is one of the strongest one in nature, while the out-of-plane π bond contributes to the electron delocalisation as well as to the electron conduction, and weak interactions between graphene layers.12,13 These characteristics are the origin of graphene extraordinary properties such as high mechanical strength, excellent electronic and thermal conductivity, tuneable band gap and large surface area.12,13 Even though the initial method utilised by Geim to isolate a graphene layer (mechanical exfoliation) is not suitable for mass production, since then several methods have been developed to produce graphene in large quantities,14,15 with chemical,16 or thermal reduction methods17 of graphene oxide being among the most popular. In the past few years, several papers have appeared either reviewing the fundamental electrochemical properties of carbon nanomaterials and/or their electrochemical applications.18 Among graphite-based materials, graphite nanoplatelets (GNPs) consist of a small stack of graphene with an average thickness of ca. 5–10 nanometers and with varying sizes up to 50 microns. This material is interesting because the short stacks of platelet-shaped graphene sheets are like those found in the walls of carbon nanotubes, but in a planar form.19 Their low cost and suitability for mass production make GNPs an interesting material for the fabrication of electrode substrates. One of the useful properties of the GNP is the easy dispersion of the flakes in benign solvents like water or ethanol without the use of surfactants.20,21 An important aspect to consider in electroanalysis is also the functionalisation of the electrode surface in order to confer high sensitivity and selectivity to the analytical device.10,22,23 In this respect, ionomers (ion-containing polymers) have been widely used for the modification of electrode surfaces.24–26 Ascorbic acid (AA), known also as vitamin C, is a molecule with important physiological roles. AA is a neuromodulator that acts as a reactive oxygen species scavenger in the brain and protects cells from oxidative stress.27,28 AA also occurs naturally in some foods and is extensively added by the food and pharmaceutical industries as a dietary supplement. The deficiency of AA has been associated with several pathologies such as infertility, mental illness, cancer and AIDS.29–32 Nowadays, the detection of AA is performed using spectroscopy,33,34 chromatography,35 photometry,36 and capillary electrophoresis37 techniques. However, these methods are complicated, time consuming and typically require pretreatment of the sample.38–40 In this contribution we report for the first time the electroanalytical applications of an anion exchange ionomer (TPQPCl)/GNPs composite-coated electrode for the simultaneous determination of ascorbic acid and dopamine. We found that by varying the ionomer/GNPs ratio it is possible to tune the selectivity towards detection of AA without interference of DA or even the possibility to detect and quantify simultaneously AA and DA under appropriate ionomer/carbon ratio conditions.
2. Material and methods
2.1 Reagents and solutions
Graphite nanoplatelets (surface modified friable nano-graphite) were obtained from Perpetuus Carbon Technologies Ltd (UK). Graphite nanoplatelets were synthesized by employing dielectric barrier discharge (DBD) plasma with various working gases.41,42 TPQPCl was synthesized by quaternary phosphorization of CMPSf with tris(2,4,6-trimethoxyphenyl)phosphine using the procedure developed by Shuang Gu et al.43 The structure of TPQPCl is shown in Scheme 1.
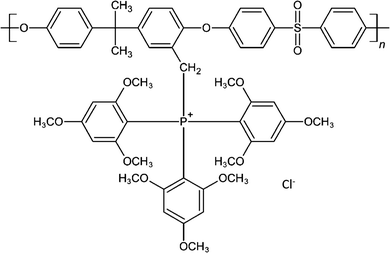 |
| | Scheme 1 Structure of tris(2,4,6-trimethoxyphenyl)polysulfone-methylene quaternary phosphonium chloride (TPQPCl). | |
The resulting TPQPCl ionomer was dried at room temperature. L-Ascorbic acid and dopamine were purchased from Sigma Aldrich. NaCl (99.5%) and ethanol (absolute HPLC grade) were purchased from Acros Organics. Aqueous solutions of NaCl at different pH values were prepared by adjusting the as-prepared solutions of NaCl with aliquots of 0.5 M HCl or 0.5 M NaOH solutions as appropriate. All aqueous solutions were prepared from doubly distilled Milli-Q reagent water (Millipore Corp.) with a resistivity of ≥18 MΩ cm at 25 °C.
2.2 Apparatus
Scanning electron microscopy (SEM) was carried out using a JEOL JEM-2011. Raman spectroscopic analysis was performed using a Renishaw inVia Raman microscope with a 532 nm excitation laser. FTIR spectrometer data were obtained using a Perkin-Elmer instrument. The zeta-potentials of the TPQPCl, GNPs and TPQPCl/GNPs composite solutions were measured using a Zetasizer (Nano-ZS, Malvern Instruments). The zeta-potential measurements were performed at room temperature in solutions at different pH values. Cyclic voltammetry measurements were performed using a CH Instrument Model 705E electrochemical work station using a conventional three-electrode cell. A glassy carbon electrode (IJ Cambria) of 3 mm diameter was used as the working electrode and a platinum wire as the counter electrode. All potentials were quoted versus an Ag/AgCl reference electrode and all the measurements were recorded at room temperature.
2.3 Preparation of TPQPCl/GNP composite solutions
Firstly, alcoholic solutions of GNPs were prepared using ethanol as a solvent. The concentration of GNPs in the solution was 1 wt%. The GNPs solutions in ethanol were sonicated for 30 minutes in order to obtain a uniform dispersion of the GNPs. Separately, a solution of the TPQPCl ionomer was prepared by dissolving 0.05 wt% of the ionomer in ethanol. The TPQPCl solution was sonicated for 10 minutes in order to obtain a homogenous solution. Then, TPQPCl/GNPs composite solutions were prepared by mixing 1 ml of the GNPs ethanolic dispersion with 1 ml of the TPQPCl ionomer solution in ethanol. The resulting TPQPCl/GNPs composite solution was sonicated for 30 min obtaining a concentration of 0.025 wt% of TPQPCl in ethanol. Different concentrations of TPQPCl were prepared by varying the amount of TPQPCl in the composite solution. Noticeably, the presence of TPQPCl in the solution allows achieving a more effective dispersion of the GNPs without agglomeration or precipitation up to 2 months after the preparation (see Fig. S1, ESI†).
2.4 Preparation of TPQPCl/GNPs coated electrodes
Glassy carbon electrodes (GCEs) were firstly polished for 5 min with different grades of alumina powder (0.1 and 0.05 μm, respectively) on a polishing disk. Then, GCEs were sonicated in ethanol and rinsed with large amounts of DI water. The coated electrodes were prepared by drop casting 2 μl of the ethanolic solution of TPQPCl/GNPs onto GCEs and dried in air. The as-prepared TPQPCl/GNPs coatings were stable up to three weeks when left in the electrolyte solutions.
2.5 Detection of ascorbic acid in real samples
The TPQPCl/GNPs coated electrodes were tested in commercial samples of orange juice and vitamin C tablets. Detection of AA was performed in samples of orange juice without any pretreatment. 0.1 M NaCl as a supporting electrolyte was added to the orange juice. The pH of the orange juice was read as pH = 4 using a HI-2002 Edge pH meter. Detection of AA in vitamin C tablets was performed by dissolving vitamin C tablets in 0.1 M NaCl solutions, and then different aliquots of these solutions were added to the electrochemical cell for the analysis.
3. Results and discussions
3.1 Microscopic and spectroscopic characterisation of TPQPCl/GNPs-coated electrodes
The morphology of the nanocomposite was analysed using SEM. A SEM picture of 2 μl TPQPCl/GNPs drop casted onto Si/SiO2 slides shows graphite flakes distributed around the whole surface. The dimensions of the flakes are ca. 70 nm in diameter (see Fig. S2, ESI†). The graphitic nature of the TPQCPl/GNPs was also proved using Raman spectroscopy. Fig. S3 (ESI†) shows the Raman spectrum related to GNPs and TPQCPl/GNPs. The Raman characteristic peaks are D and G bands corresponding to the positions 1330 cm−1 and 1575 cm−1 respectively.44,45 The D band is related to the sp3 bonds around the radial direction of the carbon atoms in the graphitic material, which is associated with the defects in the graphene flakes.46 The G band is related to the movement along the bonding direction of the carbon atoms attached by sp2 bonds in the graphitic materials. The ratio between the intensity of the two bands (ID/IG) is closely related to the grade of defects in the graphitic material. As shown in Fig. S3 (ESI†) both the Raman spectra for GNPs and TPQCPl/GNPs point out these characteristic bands and confirm the presence of graphite nanoplatelets in the composite. Furthermore, the calculated ratio ID/IG for the GNPs is 0.46, while for TPQCPl/GNPs is 1.03. The higher ratio obtained for TPQCPl/GNPs shows that the polymer covers uniformly the graphite nanoplatelets surface but also increases the defects in the composite material.47,48 This may occur through either interactions of the ionomer at the edges of the graphitic structure and intercalation between the graphitic layers. Noticeably, the polymer stabilises the graphitic dispersion avoiding the agglomeration of the graphite nanoplatelets over time.
3.2 Electrochemical characterisation of TPQPCl/GNP coated electrodes
The electrochemical behaviour of the bare GCE and TPQPCl/GNPs was studied using cyclic voltammetry (CV). The electrochemical characterisation was performed using Fe(CN)64− as a redox probe. Fig. 1 shows the CVs of a bare GCE and the TPQPCl/GNPs-coated electrode loaded in 5 mM potassium ferrocyanide solution and 0.1 M NaCl supporting electrolyte solution. Two well-defined redox peaks, related to the oxidation of Fe2+ to Fe3+ at ca. 0.2 V vs. Ag/AgCl and reduction of Fe3+ to Fe2+ at ca. 0.14 V vs. Ag/AgCl, are clearly visible for both the bare GCE and the TPQPCl/GNPs-coated electrodes.
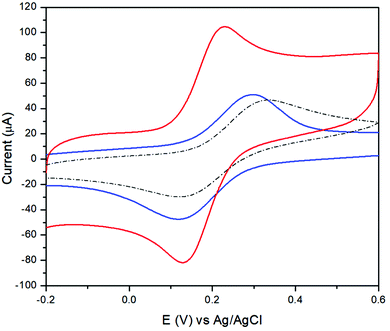 |
| | Fig. 1 CVs recorded at the bare GCE (dotted line), TPQPCl 0.025% (blue line), TPQPCl/GNPs 0.025% (red line) containing 5 mM Fe(CN)64− and 0.1 M NaCl supporting electrolyte solutions; scan rate, 100 mV s−1. | |
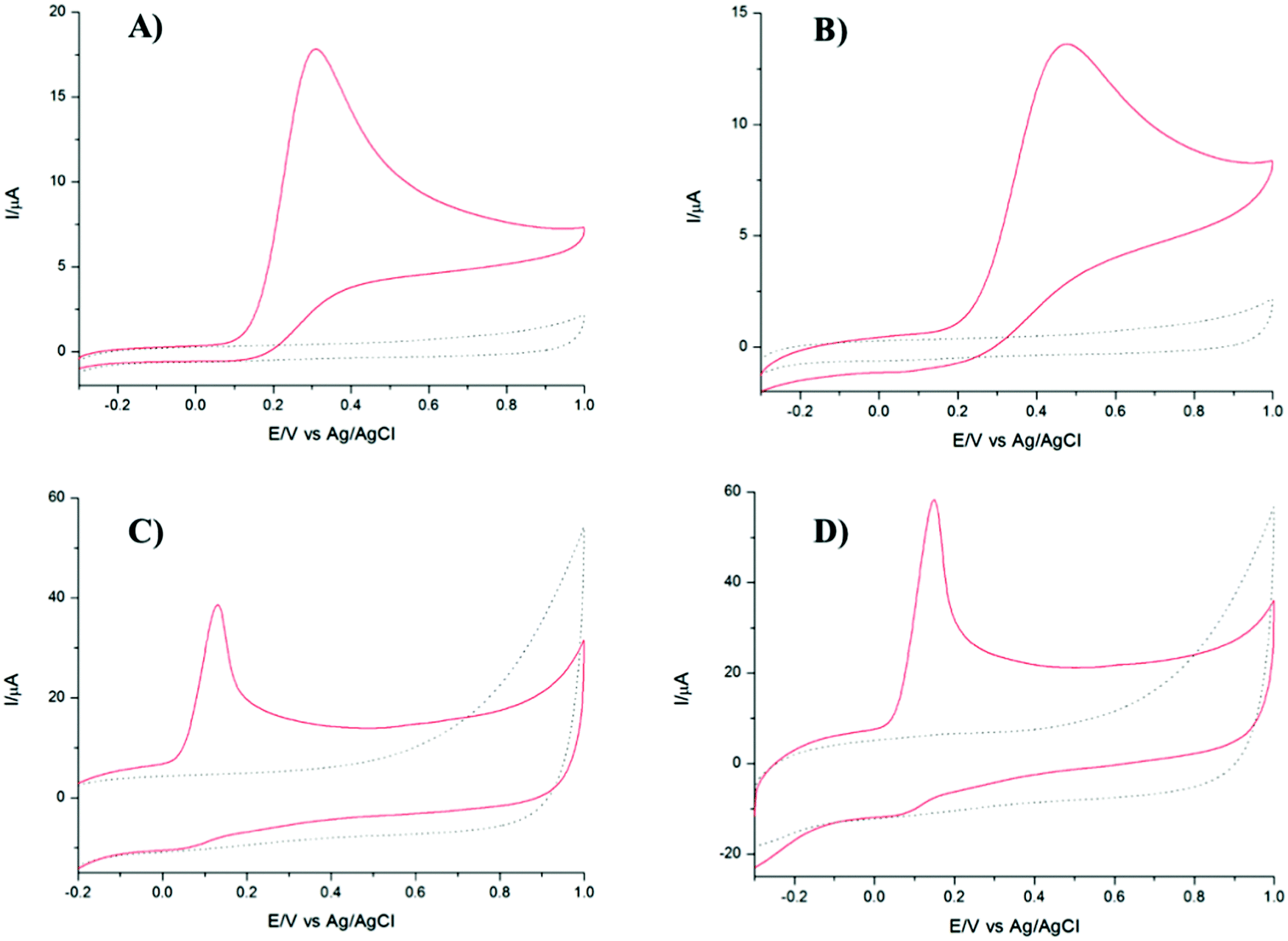
The CVs related to TPQPCl/GNPs recorded under loading conditions with a redox mediator in the electrolyte solution reveals a higher peak current and peak–peak separation, ΔEp, (with ΔEp = Efp − Ebp, where Efp and Ebp are the peak potentials for the forward and backward scans) that are slightly larger (ΔEp = 90 mV) than those expected for a Nernstian process. The CVs recorded in Fig. 2 show the redox behaviour of different coated electrodes after transferring in solution containing only the supporting electrolyte. The background currents increase with respect of the content of GNPs. This is not surprising taking into account that with the increase in the graphitic content the effective area of the electrode increases as evidenced by the higher capacitive currents observed. It is worth noting the unusual characteristics of the CVs in Fig. 2B and C in comparison with Fig. 2A and D. As expected, the ionomer TPQPCl introduces both charge and hydrophobicity. The peak currents increase linearly with the concentration of the ionomer, as a result of the preconcentration capabilities operated by the positively charged TPQPCl ionomer with the negatively charged ferro/ferricyanide redox couple. However, the CVs reported in Fig. 2B and C do not show or show only marginally the presence of peak of the redox couple compared to Fig. 2A and D. We explain this effect taking into consideration the values of the zeta potential of different ionomer/GNPs composite ratios reported in Table 1. For this experiment, we kept constant the concentration of GNPs as 0.5 wt% for all the nanocomposites. The results herein reported are compared with a standard GNPs composite solution made of the most common ionomer Nafion that, as expected, depicts negative values of the zeta potential across the whole range of pH values, from pH 2 to pH 10 due to the presence of the highly acidic sulfonated groups of the Nafion structure. Instead, the dispersion of pristine GNPs switches from a positive value of +19 at pH 2 to a negative value of −6 at pH 4, with a more negative trend as the pH increases (Table 2). Instead, the pristine TPQPCl solution reveals the positive zeta potential across all pH range values; therefore, under these conditions, it is able to preconcentrate and retain the negatively charged ferrocyanide anions. For the composite solutions utilised to obtain the coating as in Fig. 2B and C, the negative charges of GNPs (in fact GNPs possess hydroxylated and/or carboxylated functionalities) are neutralised by the positive charge of the phosphonium groups of TPQPCl, and therefore the ionomer/GNPs composite is unable to preconcentrate the negatively charged redox mediator. As the ionomer concentration increases, more positive charges are available for the incorporation of the negatively charged redox mediator, leading to the appearance of the typical redox peaks (Fig. 2A and D) of the ferro/ferricyanide redox couple. It is worth mentioning that the hydrophobicity introduced by the addition of the ionomer is important to confer stability to the composite dispersion solution.
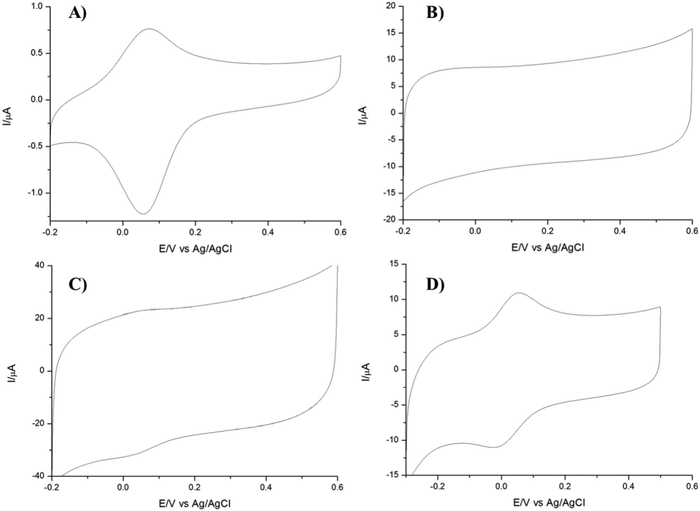 |
| | Fig. 2 CVs of (A) TPQPCl 0.025%, (B) TPQPCl/GNPs 0.025%, (C) TPQPCl/GNPs 0.5%, and (D) TPQPCl/GNPs 1.25% loaded in 5 mM Fe(CN)64− after transferring in the 0.1 M NaCl supporting electrolyte; scan rate, 50 mV s−1. | |
Table 1 Zeta potential of different ionomer/GNP composite ratios, using 0.5 wt% of GNPs
| Concentration of TPQPCl wt% |
Nafion/GNPs |
TPQPCl/GNPs |
| 0.025 |
−24.3 |
14.2 |
| 0.5 |
−32.8 |
38.3 |
| 1.25 |
−46.7 |
45.3 |
Table 2 Zeta potential of the composite material at different pH values, using 0.5 wt% of GNPs
| pH |
GNPs |
TPQPCl 0.025% |
TPQPCl/GNPs 0.025% |
Nafion/GNPs 0.5% |
| 2 |
19.53 |
32.36 |
33.73 |
−23.57 |
| 4 |
−6.62 |
27.3 |
32.2 |
−25.8 |
| 7 |
−29.23 |
21.21 |
16.97 |
−32.8 |
| 10 |
−29.77 |
19.13 |
−13.7 |
−33.4 |
3.3 Electrochemical behaviour of ascorbic acid at TPQPCl and TPQPCl/GNP coated electrodes
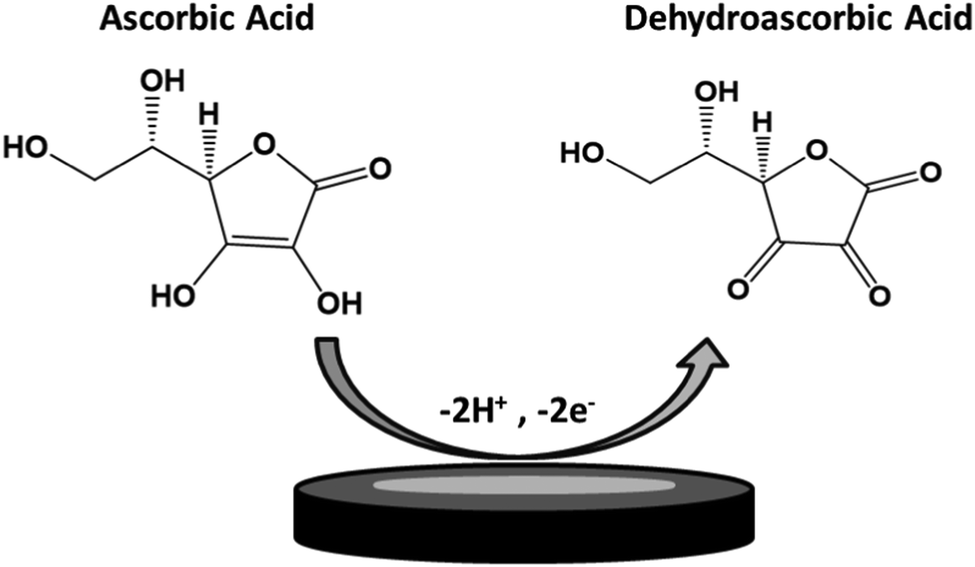
TPQPCl and TPQPCl/GNP-coated electrodes were tested towards the detection of anionic analytes, such as ascorbic acid. The electrochemical response of ascorbic acid has been recorded using a bare GCE, TPQPCl 0.025%, GNPs, and TPQPCl/GNPs 0.025% in 0.1 M NaCl solution at pH 4 in the presence of 1 mM ascorbic acid. Fig. 3 shows the CVs of ascorbic acid at different coated electrodes. All the electrodes show an oxidation peak related to the irreversible oxidation of ascorbic acid to dehydroascorbic acid according to Scheme 2.49
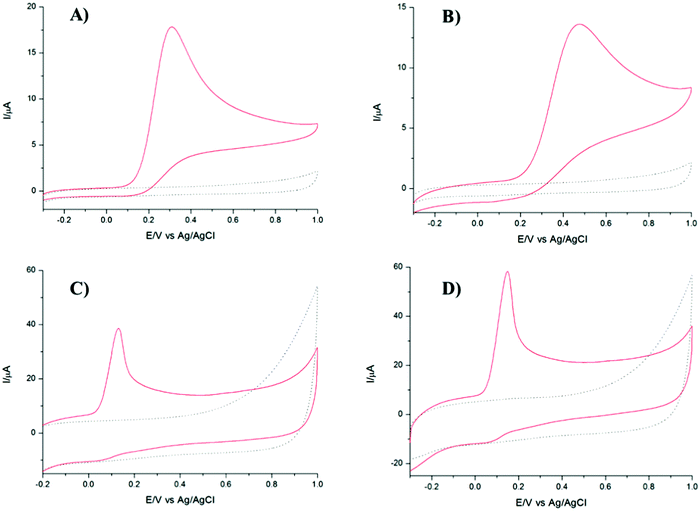 |
| | Fig. 3 CVs of (A) bare GCE, (B) TPQPCl 0.025%, (C) GNPs, and (D) TPQPCl/GNPs 0.025% recorded in 1 mM AA and 0.1 M NaCl supporting electrolyte solutions (pH 4); scan rate: 50 mV s−1. The dotted curves represent the CVs (background currents) recorded in the 0.1 M NaCl supporting electrolyte only. | |
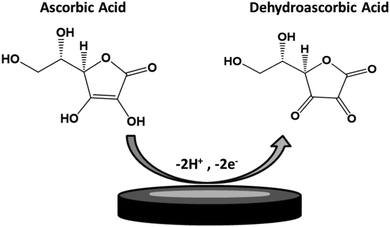 |
| | Scheme 2 Mechanism of oxidation of ascorbic acid to dehydroascorbic acid. | |
Noticeably, the intensity of peak currents increases from 18 μA to 59 μA for the TPQPCl/GNPs 0.025% modified electrodes as an indication of the higher sensitivity achieved by GNPs. The peak current for the TPQPCl 0.025% modified electrode is slightly lower than that of the bare GCE due to the lower diffusion coefficient of ascorbic acid in the ionomer-coated electrode. Also, the peak potential shifts to a more positive potential from 0.3 V to 0.47 V in the case of the TPQPCl 0.025% modified electrode. This effect is ascribed to the fact that the preconcentration capability of TPQPCl causes a shift to a more positive potential of oxidation due to the slower diffusion of ascorbic acid within the pristine TPQPCl coated film. Interestingly, the addition of GNPs to the composite solution causes a significant shift towards negative potentials from 0.3 V to 0.14 V vs. Ag/AgCl, along with a 4-fold increase of the peak current intensity. This peak potential shift observed in the TPQPCl/GNPs 0.025% is attributed to the presence of C–OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO residues in the graphite nanoplatelets in agreement with the work of Zanardi et al.50 Also, the addition of GNPs leads to an increase of the background current as an indication of the increased surface active area of the electrode. These results point out to the beneficial effect operated by the addition of GNPs to the ionomer solution. The substantial increase of the peak current values with the GNPs loading can be explained with the electrical conductivity of GNPs, as well as its high surface area, high electrical conductivity and increased porosity, which facilitate the diffusion of the electroactive species in comparison with the pristine TPQPCl coated film.
O and O–CO residues in the graphite nanoplatelets in agreement with the work of Zanardi et al.50 Also, the addition of GNPs leads to an increase of the background current as an indication of the increased surface active area of the electrode. These results point out to the beneficial effect operated by the addition of GNPs to the ionomer solution. The substantial increase of the peak current values with the GNPs loading can be explained with the electrical conductivity of GNPs, as well as its high surface area, high electrical conductivity and increased porosity, which facilitate the diffusion of the electroactive species in comparison with the pristine TPQPCl coated film.
Fig. S4 (ESI†) illustrates the effect of the scan rate on both TPQPCl and TPQPCl-coated electrodes, in the presence of 1 mM ascorbic acid and 0.1 M NaCl supporting electrolyte solutions (pH 4). The values of the oxidation potential were found to be larger up to 244 mV at 1 V s−1, whereas they decreased down to ca. 120 mV at 5 mV s−1. The peak current increases linearly with the square root of the scan rate from 10 mV s−1 to 1 V s−1, indicating a diffusion-controlled process. Note that on the time scale of the experiment (approximately 15 min to run a complete set of CVs), the loss of the redox mediator as evidenced from CVs recorded at the same scan rate at the beginning and end of the measurements was typically less than 15%.
3.4 Dependence of detection of ascorbic acid on pH
In order to ascertain whether the voltammetric signal is pH dependent, several LSVs were recorded at different ranges of pH from pH 2 to pH 11. The results obtained showed a strong dependence of the voltammetric signal from the pH. TPQPCl modified electrodes (Fig. 4A) show little dependence from the pH in terms of peak current intensity; however, the irreversible oxidation of AA shifts from 55 mV at pH 2 to 31 mV at pH 11, with the peak potential becoming less positive as the pH increases. This situation was markedly different from the TPQPCl/GNPs-modified electrodes. While the peak potential for the oxidation of AA appears at a lower potential similarly to the TPQPCl-coated electrodes, the intensity of the peak current decreases dramatically as the pH increases, until it almost disappears at very alkaline values of pH. We explain this behaviour with the fact that at alkaline pH and as the concentration of GNPs increases, there is an excess of the negative charge functionalities of GNPs in the form of carboxylated (COO−) GNPs which cause repulsion with the negative charges of AA (pKa 4.2).51 This effect can be justified with the negative values of the zeta potential for the TPQPCl/GNPs composite at pH higher than 7 (Table 2). Instead, at acidic pH values the GNPs are present in the undissociated form (C–OH, CO and O–CO) without electrostatic repulsion with AA. The measurements were performed in 1 mM AA and 0.1 M NaCl and afterwards the pH was changed by adding NaOH or HCl as appropriate. It has been found that the highest oxidation current peak for AA was at pH 4.52
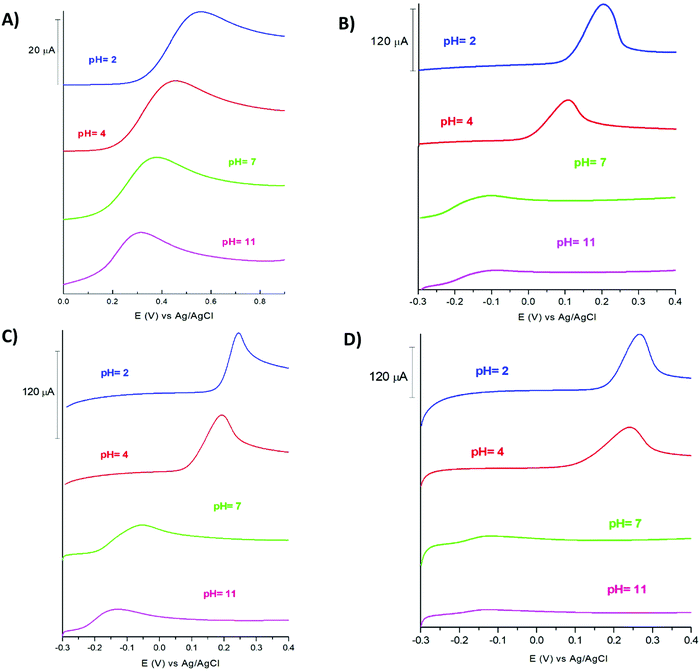 |
| | Fig. 4 LSV of (A) TPQPCl 0.025%, (B) TPQPCl/GNPs 0.025%, (C) TPQPCl/GNPs 0.5%, and (D) TPQPCl/GNPs 1.25% coated electrodes recorded in 1 mM AA and 0.1 M NaCl supporting electrolyte solutions at various pH values. Scan rate, 50 mV s−1. | |
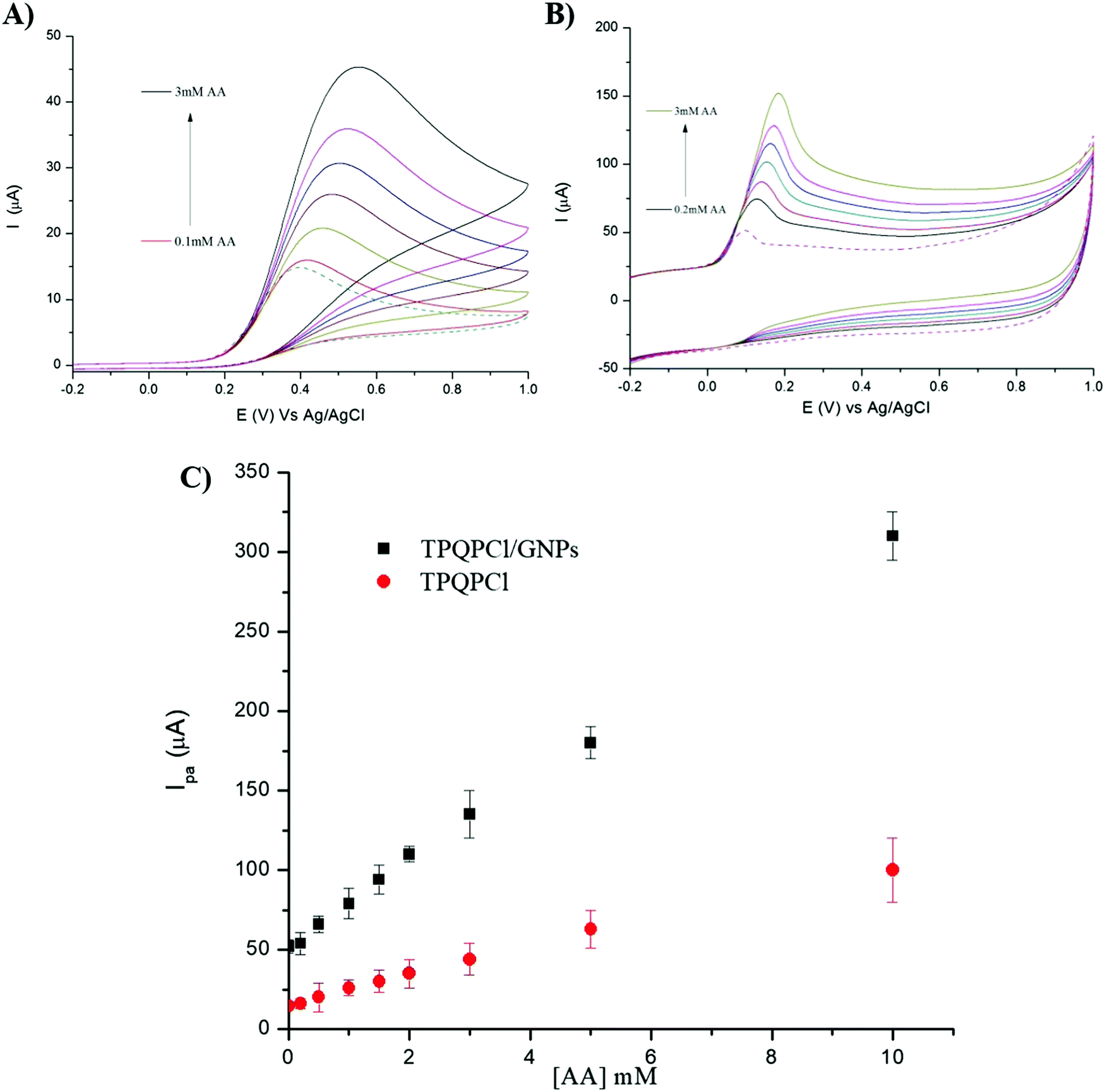
3.5 Detection limit of ascorbic acid
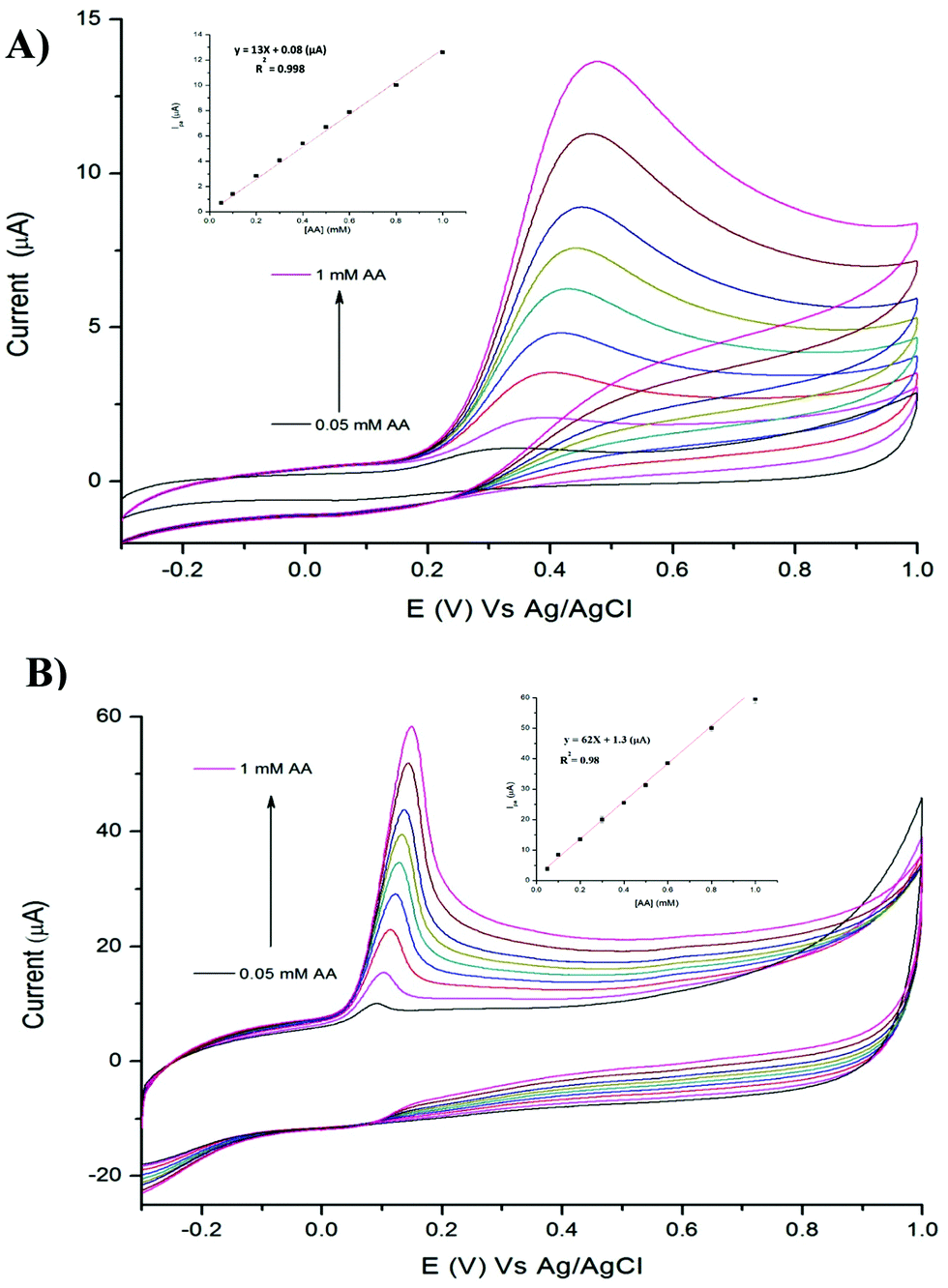
We investigated the possibility to utilise TPQPCl and TPQPCl/GNPs-coated electrodes for the preconcentration and detection of a typical anionic analyte such as ascorbic acid (AA). Fig. 5 shows the CVs recorded in the 0.1 M NaCl solution (pH 4) supporting electrolyte with increasing concentrations of AA (from 0.05 mM to 1 mM) for TPQPCl and TPQPCl/GNPs 0.025 wt% coated electrodes. Both CVs show the typical oxidation peak related to the irreversible oxidation of ascorbic acid to dehydroascorbic acid. Noticeably, the peak current recorded at the ionomer/GNPs composite is ca. 4-fold higher than the peak current at the ionomer-coated electrode only. Significantly, the potential of oxidation at the TPQPCl/GNPs-coated electrode (Fig. 5B) occurs at 145 mV compared to 470 mV observed for the pristine TPQPCl-coated electrode (Fig. 5A). This confirms the beneficial effect of GNPs in the composite film which facilitates the electrochemical oxidation of AA with a concomitant increase of the peak current. Note, again, that the addition of GNPs causes an increase of the background currents as a result of the high electrode surface active area. Despite this effect, the TPQPCl/GNPs-coated electrodes show higher sensitivity than TPQPCl-coated electrodes. For both electrodes, the peak currents increase linearly with the concentration of AA in the range of 0.05 mM–1 mM (see the inset of Fig. 5A and B). The limits of detection (LoDs) were calculated using the 3Sb/m method where Sb is the standard deviation obtained from 3 measurements of the blank signal and m is the slope obtained from the calibration plot.45 The LoD values of AA are calculated as 3.5 μM and 4.8 μM for TPQPCl and TPQPCl/GNPs, respectively. The sensitivity values are determined to be 0.18 μA μM−1 cm−2 for the TPQPCl and 0.88 μA μM−1 cm−2 for the TPQPCl/GNPs composite-coated electrode.
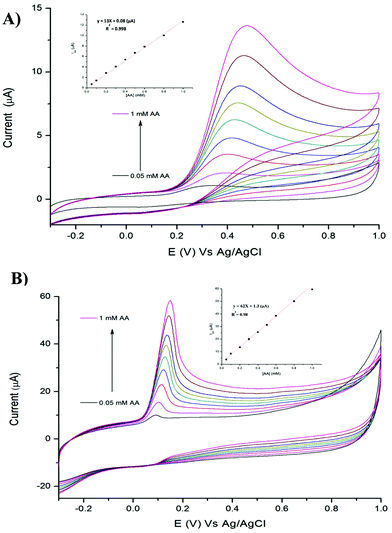 |
| | Fig. 5 CVs of (A) TPQPCl, and (B) TPQPCl/GNPs at different concentrations of AA from 0.05 mM to 1 mM recorded in the 0.1 M NaCl supporting electrolyte (pH 4); scan rate, 50 mV s−1. Inset: Plot of anodic peak currents vs. concentration of AA. | |
3.6 Simultaneous and selective determination of ascorbic acid and dopamine
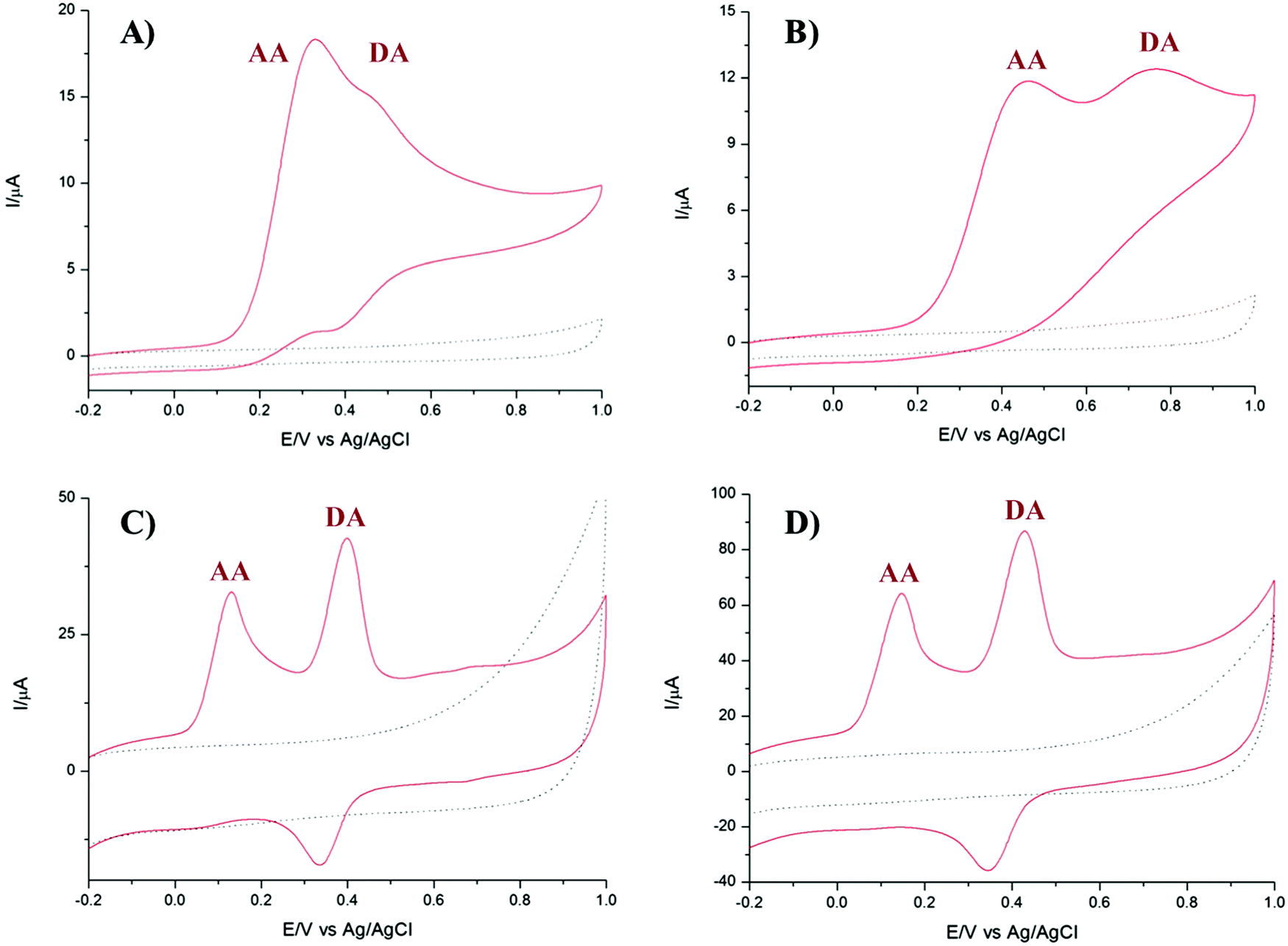
It is well known that dopamine (DA) and AA oxidise at similar potentials, and hence simultaneous detection of these species is problematic. In biological fluids, detection of DA is affected by the interference of AA, whose concentration is well in excess compared to DA. Here we investigated the voltammetric behaviour of AA in the presence of DA. Fig. 6 shows the CVs of the bare GCE, TPQPCl, GNPs and TPQPCl/GNPs 0.025 wt% in the presence of 1 mM AA and 0.1 mM DA dissolved in the 0.1 M NaCl supporting electrolyte (pH 4) at a scan rate of 50 mV s−1. It is evident from Fig. 6A the overlap of the oxidation peaks of AA and DA at around 0.4 V, which corresponds to the irreversible oxidation of AA and DA occurring at the bare GCE. Therefore, as expected, the simultaneous determination of both analytes is very difficult at the bare GCE.53,54 This effect is similar in the TPQPCl-coated electrode (Fig. 6B) where, again, the oxidation peaks of AA and DA are very close each other, which make difficult the simultaneous detection of both analytes. In contrast, the CVs recorded at GNPs and TPQPCl/GNPs-coated electrodes show the presence of two well defined peaks at 0.14 mV and 0.43 mV, for AA and DA, respectively. The difference between the voltammetric behaviour of GNPs (Fig. 6C) and TPQPCl/GNPs (Fig. 6D) is noticeable, with the intensity of TPQPCl/GNPs about 2-fold higher compared to the GNPs-coated electrode, even though the peak separation for TPQPCl/GNPs is slightly larger (0.02 V). The difference in the intensity of the peak current is even more pronounced (up to 5-fold higher) when the TPQPCl/GNPs-coated electrode (Fig. 6D) is compared with TPQPCl (Fig. 6B). These results show that the TPQPCl/GNPs electrodes exhibit enhanced electrocatalytic activity for the oxidation of AA and DA. As explained previously, GNPs have excellent electrical transport properties which can effectively increase the rate of electron transfer at the interface of carbon electrodes.
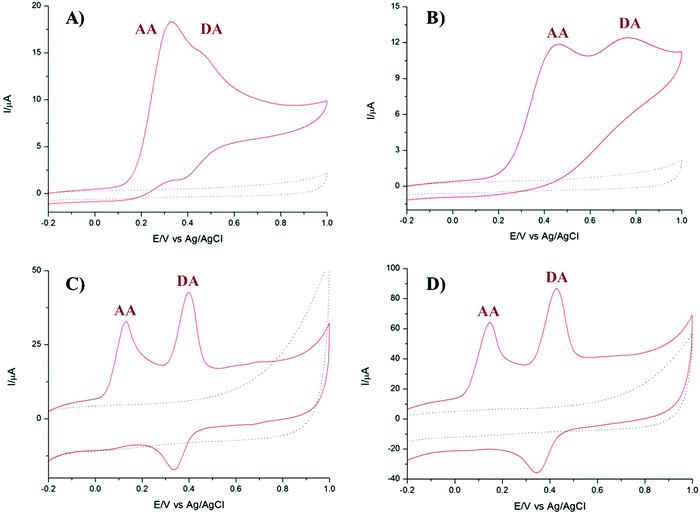 |
| | Fig. 6 CVs of (A) bare GCE, (B) TPQPCl, (C) GNPs, and (D) TPQPCl/GNPs recorded in 1 mM AA and 0.1 mM DA in the 0.1 M NaCl supporting electrolyte (pH 4); scan rate, 50 mV s−1. The dotted curves represent the corresponding CV (background current) recorded in only the supporting electrolyte. | |
To investigate further the selectivity of TPQPCl/GNPs-coated electrodes, we performed the detection of AA in the presence of different concentrations of dopamine. Fig. 7 shows the CVs for the oxidation of AA where the concentration of AA is maintained constant at 1 mM, whereas the concentration of DA increases from 0.1 mM to 2 mM in 0.1 M NaCl (pH 4). The peak current of DA increases with the DA concentration, whereas, as expected, the peak current of AA slightly decreases. Note that the concentrations of DA herein utilised are very high when compared to the typical concentrations of DA encountered in extracellular fluids (typically in the order of tens of nanomolar), with the concentration of AA well in excess (up to a factor 103).55
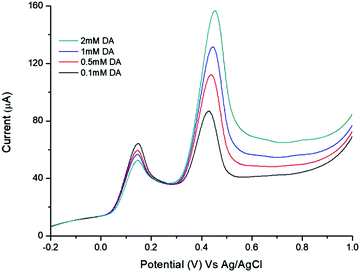 |
| | Fig. 7 LSV of the TPQPCl/GNP-coated electrode recorded in 0.1 M NaCl supporting electrolyte (pH 4) containing a constant concentration of AA (1 mM) and different concentrations of DA from 0.1 mM to 2 mM. Scan rate, 50 mV s−1. | |
These results point out how the TPQPCl/GNPs-coated electrodes are sensitive towards the detection of each analyte and allow the simultaneous detection of AA and DA without apparent interference.
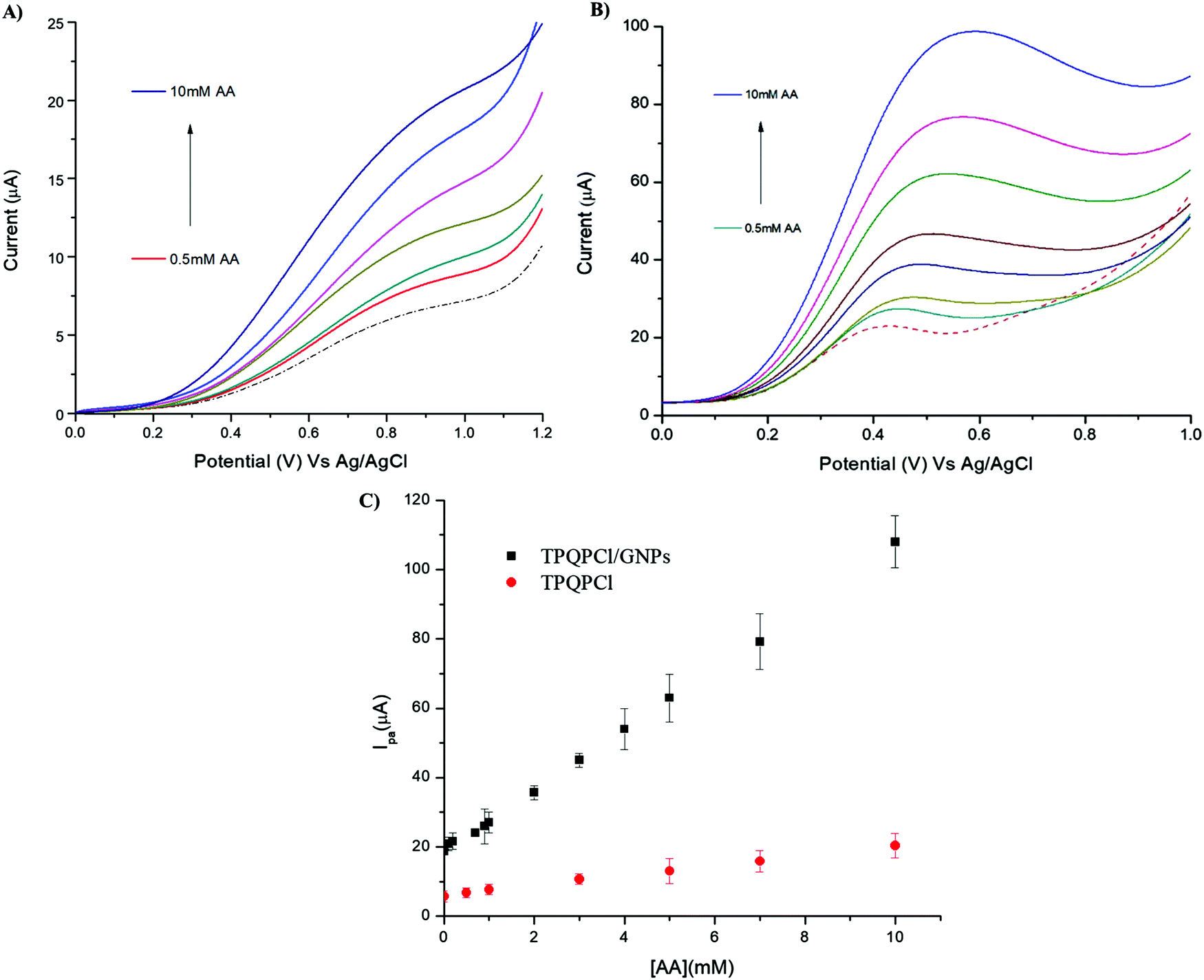
3.7 Detection of ascorbic acid in real samples
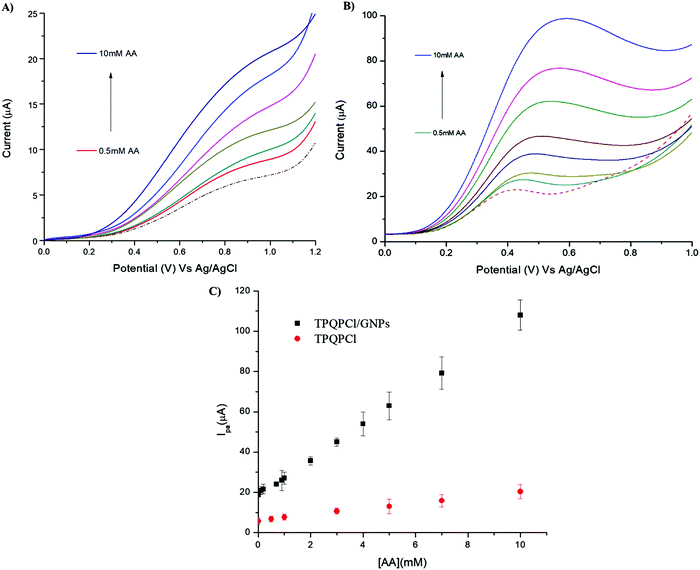
The as-prepared TPQPCl/GNPs-coated electrodes were utilised for the detection of AA in two different real samples, such as vitamin C tablets and orange juice. In the case of the vitamin C tablets, the effervescent tablet was dissolved in 0.1 M NaCl (pH 4) and different aliquots of this solution were added in the electrochemical cell. Fig. 8 shows the voltammetric behaviour recorded at the TPQPCl (Fig. 8A) and TPQPCl/GNPs-coated electrode (Fig. 8B) in the 0.1 M NaCl supporting electrolyte. The CVs show the typical irreversible oxidation peak of AA with the oxidation potential being shifted to more negative values (ca. 250 mV) in the case of the TPQPCl/GNPs-coated electrode as previously observed. In both cases, the peak current increases linearly with the concentration of AA from 0.1 mM to 10 mM. The calculated LoD is 10.8 μM for TPQPCl/GNPs.
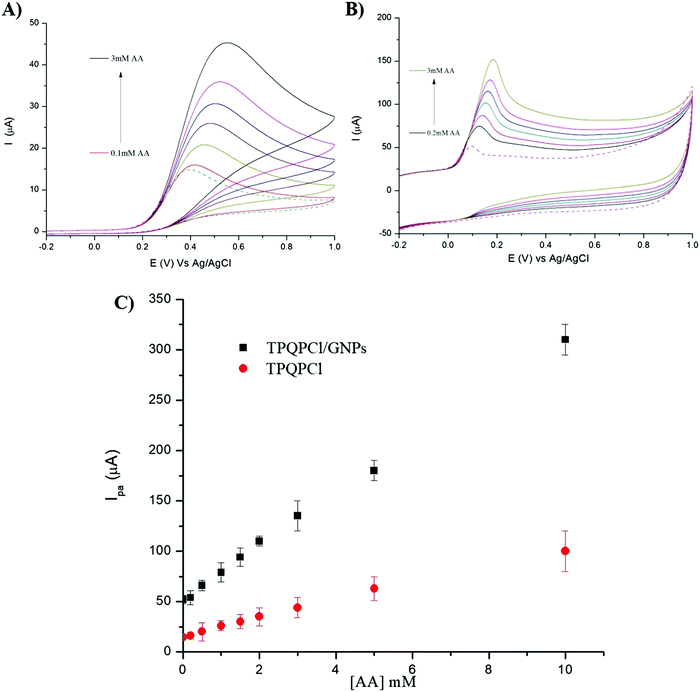 |
| | Fig. 8 CVs of (A) TPQPCl, and (B) TPQPCl/GNPs of vitamin C tablet (dotted line) and after the addition of various concentrations of AA from 1 mM to 3 mM. Supporting electrolyte, 0.1 M NaCl (pH 4); scan rate, 50 mV s−1. (C) Plot of oxidation currents vs. concentration of AA (from 1 mM to 10 mM) for TPQPCl (red) and TPQPCl/GNPs (black). | |
In the case of the sample of orange juice, the detection of AA was more difficult. Carton orange juice was utilised without any pretreatment (no dilution, nor filtration or adjustment of pH) apart from the addition of 0.1 M NaCl as the supporting electrolyte. The pH of the orange juice was read as pH = 3.6. Fig. 9 shows the CVs of the orange juice before and after the addition of different concentrations of AA. The CV related to TPQPCl is very different compared to the situation previously observed in the vitamin C sample. The intensity of the oxidation peak current is lower compared to that observed for the vitamin C sample and also the shape of the peak is drastically different. In the case of the TPQPCl/GNPs-coated electrode, and compared to the CVs recorded for the vitamin C samples, the intensity of the peak current is drastically reduced (more than 2-fold lower), as well as both the shape and the value of the oxidation peak potential show a rather different behaviour. In the sample of orange juice the peak is much broader and the oxidation value shifts to a more positive potential (up to ca. 400 mV for TPQPCl and 250 mV for TPQPCl/GNPs) compared to vitamin C. It is well known that ascorbic acid is prone to fast oxidation when in contact with air, and therefore we did not perform dilution or filtration of the samples whatsoever, using orange juice immediately as opened. Having utilised orange juice samples without any pretreatment, both the TPQPCl and TPQPCl/GNP electrodes are more susceptible to fouling and matrix effects that strongly affect the voltammetric analysis. However, the plot of the peak current vs. AA concentration shows a linear response from 0.5 mM to 10 mM and a calculated limit of detection of 67 μM for the TPQPCl/GNPs-coated electrode. Noticeably, the TPQPCl/GNPs-coated electrode results to be more resistant to these matrix effects compared to the pristine TPQPCl electrode.
 |
| | Fig. 9 LSV of (A) TPQPCl, and (B) TPQPCl/GNPs-coated electrodes recorded in commercial orange juice (dotted line) and after the addition of various concentrations of AA from 0.5 mM to 10 mM AA. Supporting electrolyte, 0.1 M NaCl (pH 4) and scan rate, 50 mV s−1. (C) Plot of oxidation peak vs. concentration of AA for TPQPCl (red) and TPQPCl/GNPs (black). | |
In both analysis (vitamin C and orange juice), the recovery values were in the range of 96% indicating the suitability of TPQPCl/GNPs-coated electrodes for the detection of AA in real samples. We point out the fact that even though our system does not show improvement in terms of limit of detection compared to other studies reported in the literature (see Table 3), instead, the wide linear range response allowed the determination of AA in real samples without any pretreatment or dilution of the samples. This is certainly an advantage in the case of determination of AA in food and tablets where the concentration of AA is relatively high (mM range) to often require several stages of dilution of samples in order to prevent fouling of the electrode. However, these treatments are time consuming and cause oxidation of AA.
Table 3 Comparison of different modified electrodes for the determination of ascorbic acid as reported in the latest literature
| Modified electrode |
Linear range (μM) |
Detection limit (μM) |
Detection method |
Ref. |
| PtAu hybrid film/GCE |
69–1384 |
— |
CV |
56
|
| ZnO/RM hybrid film-coated GCE |
15–240 |
1.4 |
CV |
57
|
| MgO nanobelts/GCE |
2.5–15, 25–150 |
0.2 |
CV |
58
|
| Poly(caffeic acid) |
20–1000 |
7 |
CV |
59
|
| Edge plane pyrolytic graphite electrode (EPPGE) |
200–2200 |
71 |
CV |
60
|
| Thionine/KB-GCE |
25–5000 |
25 |
CV |
61
|
| PPy/GR/GC |
0.5–10 |
0.1 |
CV |
62
|
| Graphene–chit/GCE |
5–200 |
— |
CV |
63
|
| β-Cyclodextrin/graphene/GCE |
0.009–13 |
0.005 |
CV |
64
|
| Graphene/carbon fibre/GCE |
8–100 |
2 |
CV |
65
|
| TPQPCl/GCE |
3.5–10000 |
3.5 |
CV |
This work |
| TPQPCl/GNPs/GCE |
4.8–10000 |
4.8 |
CV |
This work |
4. Conclusions
A novel amperometric sensor for the detection of ascorbic acid in real samples (orange juice and vitamin C tablets) based on the TPQPCl/GNP composite was demonstrated. The electrochemical characterization of the TPQPCl/GNPs composite was performed using cyclic voltammetry. The electrochemical characterization reveals that TPQPCl/GNPs-coated electrodes lead to a significant increase of the oxidation peak and at a more favourable potential, showing an excellent electron transfer rate towards the oxidation of AA. This new sensor is characterised by a rapid response, high sensitivity, good reproducibility and excellent selectivity. Most importantly, we proved that simultaneous detection of dopamine without interference of ascorbic acid is possible by increasing the GNP concentration in the composite solution. Finally, the suitability of the as-prepared TPQPCl/GNPs-composite solution has been verified in real samples of orange juice and vitamin C, evidencing the substantial potential of the TPQPCl/GNPs composite solution towards detection of AA with a limit of detection of 4.8 μM, while for TPQPCl the limit of detection is 3.5 μM. Finally, the as-prepared TPQPCl/GNPs-coated electrodes have been tested for the detection of samples of untreated orange juice and vitamin C. The TPQPCl/GNPs-coated electrodes are a promising sensing platform for easy and fast detection of AA.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Engineering and Physical Sciences Research Council (EPSRC), grant EP/L013797/1. S. H.-A. gratefully acknowledges financial support from the Knowledge Economy Skills Scholarships (KESS2) under the Welsh Government's European Social Fund (ESF) convergence programme for West Wales and the Valleys. The authors are thankful to Dr Darren Oatley-Radcliffe and Dr Paul Williams for the use of the zeta potential apparatus.
References
- D. Grieshaber, R. MacKenzie, J. Voros and E. Reimhult, Sensors, 2008, 8, 1400–1458 CrossRef CAS.
- M. Pumera, A. Ambrosi, A. Bonanni, E. L. K. Chng and H. L. Poh, TrAC, Trends Anal. Chem., 2010, 29, 954–965 CrossRef CAS.
- P. Leonard, S. Hearty, J. Brennan, L. Dunne, J. Quinn, T. Chakraborty and R. O'Kennedy, Enzyme Microb. Technol., 2003, 32, 3–13 CrossRef CAS.
- P. D. Patel, TrAC, Trends Anal. Chem., 2002, 21, 96–115 CrossRef CAS.
- J. R. Windmiller and J. Wang, Electroanalysis, 2013, 25, 29–46 CrossRef CAS.
- A. Qureshi, W. P. Kang, J. L. Davidson and Y. Gurbuz, Diamond Relat. Mater., 2009, 18, 1401–1420 CrossRef CAS.
- I. Svancara, K. Vytras, K. Kalcher, A. Walcarius and J. Wang, Electroanalysis, 2009, 21, 7–28 CrossRef CAS.
- J. J. Gooding, Electrochim. Acta, 2005, 50, 3049–3060 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- Y. Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. H. Lin, Electroanalysis, 2010, 22, 1027–1036 CrossRef CAS.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178–1271 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- W. F. Chen, L. F. Yan and P. R. Bangal, Carbon, 2010, 48, 1146–1152 CrossRef CAS.
- K. R. Ratinac, W. R. Yang, J. J. Gooding, P. Thordarson and F. Braet, Electroanalysis, 2011, 23, 803–826 CrossRef CAS.
- A. Nieto, D. Lahiri and A. Agarwal, Carbon, 2012, 50, 4068–4077 CrossRef CAS.
- M. Rashad, F. Pan, A. Tang and M. Asif, Prog. Nat. Sci.: Mater. Int., 2014, 24, 101–108 CrossRef CAS.
- Y. Geng, S. J. Wang and J.-K. Kim, J. Colloid Interface Sci., 2009, 336, 592–598 CrossRef CAS PubMed.
- W. R. Yang, P. Thordarson, J. J. Gooding, S. P. Ringer and F. Braet, Nanotechnology, 2007, 18, 12 Search PubMed.
- Y. P. Song, M. Feng and H. B. Zhan, Prog. Chem., 2012, 24, 1665–1673 CAS.
- M. Pesavento, G. Alberti and R. Biesuz, Anal. Chim. Acta, 2009, 631, 129–141 CrossRef CAS.
- K. Z. Brainina, N. A. Malakhova and N. Y. Stozhko, Fresenius' J. Anal. Chem., 2000, 368, 307–325 CrossRef CAS.
- P. Ugo, L. M. Moretto and F. Vezza, ChemPhysChem, 2002, 3, 917–925 CrossRef CAS.
- B. N. Ames, M. K. Shigenaga and T. M. Hagen, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 7915–7922 CrossRef CAS.
- R. Cozzi, R. Ricordy, T. Aglitti, V. Gatta, P. Perticone and R. DeSalvia, Carcinogenesis, 1997, 18, 223–228 CrossRef CAS.
- O. Arrigoni and M. C. De Tullio, Biochim. Biophys. Acta, Gen. Subj., 2002, 1569, 1–9 CrossRef CAS.
- Y. C. Bingcheng Su, X. Yang, J. Han, H. Jia, P. Jing and Y. Wang, Int. J. Electrochem. Sci., 2017, 12, 6417–6427 Search PubMed.
- P. Shakkthivel and S.-M. Chen, Biosens. Bioelectron., 2007, 22, 1680–1687 CrossRef CAS.
- S.-A. Jung, D.-H. Lee, J.-H. Moon, S.-W. Hong, J.-S. Shin, I. Y. Hwang, Y. J. Shin, J. H. Kim, E.-Y. Gong, S.-M. Kim, E. Y. Lee, S. Lee, J. E. Kim, K.-p. Kim, Y. S. Hong, J. S. Lee, D.-H. Jin, T. Kim and W. J. Lee, Free Radicals Biol. Med., 2016, 95, 200–208 CrossRef CAS PubMed.
- J. F. Y. Fong, S. F. Chin and S. M. Ng, Biosens. Bioelectron., 2016, 85, 844–852 CrossRef CAS PubMed.
- M. R. Moghadam, S. Dadfarnia, A. M. H. Shabani and P. Shahbazikhah, Anal. Biochem., 2011, 410, 289–295 CrossRef CAS PubMed.
- Y. Michotte, M. Moors, D. Deleu, P. Herregodts and G. Ebinger, J. Pharm. Biomed. Anal., 1987, 5, 659–664 CrossRef CAS PubMed.
- T. Tono and S. Fujita, Agric. Biol. Chem., 1981, 45, 2947–2949 CAS.
- B. Habibi, M. Jahanbakhshi and M. Pournaghi-Azar, Electrochim. Acta, 2011, 56, 2888–2894 CrossRef CAS.
- M. Liu, Q. Chen, C. Lai, Y. Zhang, J. Deng, H. Li and S. Yao, Biosens. Bioelectron., 2013, 48C, 75–81 CrossRef PubMed.
- Y. Zhao, Y. Gao, D. Zhan, H. Liu, Q. Zhao, Y. Kou, Y. Shao, M. Li, Q. Zhuang and Z. Zhu, Talanta, 2005, 66, 51–57 CrossRef CAS PubMed.
- D. Zhao, G. Yu, K. Tian and C. Xu, Biosens. Bioelectron., 2016, 82, 119–126 CrossRef CAS PubMed.
- Q. Zhou, Z. Zhao, Y. Chen, H. Hu and J. Qiu, J. Mater. Chem., 2012, 22, 6061–6066 RSC.
- K. Krishnamoorthy, A. Ananth, Y. S. Mok and S. J. Kim, Sci. Adv. Mater., 2014, 6, 349–353 CrossRef CAS.
- S. Gu, R. Cai, T. Luo, Z. Chen, M. Sun, Y. Liu, G. He and Y. Yan, Angew. Chem., Int. Ed., 2009, 48, 6499–6502 CrossRef CAS PubMed.
- M. Rashad, F. Pan, A. Tang, Y. Lu, M. Asif, S. Hussain, J. She, J. Gou and J. Mao, J. Magnesium Alloys, 2013, 1, 242–248 CrossRef CAS.
- B. Dinesh, R. Saraswathi and A. Senthil Kumar, Electrochim. Acta, 2017, 233, 92–104 CrossRef CAS.
- A. Eckmann, A. Felten, A. Mishchenko, L. Britnell, R. Krupke, K. S. Novoselov and C. Casiraghi, Nano Lett., 2012, 12, 3925–3930 CrossRef CAS PubMed.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- L. G. Cançado, A. Jorio, E. H. M. Ferreira, F. Stavale, C. A. Achete, R. B. Capaz, M. V. O. Moutinho, A. Lombardo, T. S. Kulmala and A. C. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef PubMed.
- S. Pruneanu, A. R. Biris, F. Pogacean, C. Socaci, M. Coros, M. C. Rosu, F. Watanabe and A. S. Biris, Electrochim. Acta, 2015, 154, 197–204 CrossRef CAS.
- G. Maccaferri, C. Zanardi, Z. Y. Xia, A. Kovtun, A. Liscio, F. Terzi, V. Palermo and R. Seeber, Carbon, 2017, 120, 165–175 CrossRef CAS.
- F. Gao, X. Cai, X. Wang, C. Gao, S. Liu, F. Gao and Q. Wang, Sens. Actuators, B, 2013, 186, 380–387 CrossRef CAS.
- D. Zheng, J. Ye, L. Zhou, Y. Zhang and C. Yu, J. Electroanal. Chem., 2009, 625, 82–87 CrossRef CAS.
- H. Zhao, Y. Zhang and Z. Yuan, Analyst, 2001, 126, 358–360 RSC.
- L. Zhang and X. Lin, Anal. Bioanal. Chem., 2005, 382, 1669–1677 CrossRef CAS PubMed.
- R. M. Wightman, L. J. May and A. C. Michael, Anal. Chem., 1988, 60, 769A–779A CrossRef CAS PubMed.
- S. Thiagarajan and S.-M. Chen, Talanta, 2007, 74, 212–222 CrossRef CAS PubMed.
- C.-F. Tang, S. A. Kumar and S.-M. Chen, Anal. Biochem., 2008, 380, 174–183 CrossRef CAS PubMed.
- M. Li, W. Guo, H. Li, W. Dai and B. Yang, Sens. Actuators, B, 2014, 204, 629–636 CrossRef CAS.
- W. Ren, H. Q. Luo and N. B. Li, Biosens. Bioelectron., 2006, 21, 1086–1092 CrossRef CAS PubMed.
- F. Wantz, C. E. Banks and R. G. Compton, Electroanalysis, 2005, 17, 1529–1533 CrossRef CAS.
- H. Cheng, X. Wang and H. Wei, Anal. Chem., 2015, 87, 8889–8895 CrossRef CAS PubMed.
- Z. Zhuang, J. Li, R. Xu and D. Xiao, Int. J. Electrochem. Sci., 2011, 6, 2149–2161 CAS.
- Y. Wang, Y. Li, L. Tang, J. Lu and J. Li, Electrochem. Commun., 2009, 11, 889–892 CrossRef CAS.
- L. Tan, K.-G. Zhou, Y.-H. Zhang, H.-X. Wang, X.-D. Wang, Y.-F. Guo and H.-L. Zhang, Electrochem. Commun., 2010, 12, 557–560 CrossRef CAS.
- Z. Mingfang, Z. Changqing and Y. Jianshan, Electroanalysis, 2011, 23, 907–914 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tc04967j |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Robert B.
Kaspar
b,
Michael P.
Letterio
b,
Afshin
Tarat
c,
Yushan
Yan
a,
Robert B.
Kaspar
b,
Michael P.
Letterio
b,
Afshin
Tarat
c,
Yushan
Yan