
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling excimer formation in indolo[3,2,1-jk]carbazole/9H-carbazole based host materials for RGB PhOLEDs†
Chenyang
Zhao‡
 ab,
Thomas
Schwartz‡
c,
Berthold
Stöger
d,
Fraser J.
White
e,
Jiangshan
Chen
*af,
Dongge
Ma
af,
Johannes
Fröhlich
c and
Paul
Kautny
*c
ab,
Thomas
Schwartz‡
c,
Berthold
Stöger
d,
Fraser J.
White
e,
Jiangshan
Chen
*af,
Dongge
Ma
af,
Johannes
Fröhlich
c and
Paul
Kautny
*c
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China
bUniversity of Science and Technology of China, Hefei, 230026, China
cInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163, A-1060 Vienna, Austria. E-mail: paul.kautny@tuwien.ac.at
dX-Ray Centre, TU Wien, Getreidemarkt 9, A-1060 Vienna, Austria
eRigaku Oxford Diffraction, Unit B6 Chaucer Business Park, Watery Lane, Kemsing, Sevenoaks, UK
fInstitute of Polymer Optoelectronic Materials and Devices, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou, 510640, China. E-mail: msjschen@scut.edu.cn
First published on 31st August 2018
Abstract
Three novel materials (5CzICz, Cz2ICz and Cz3ICz), based on the indolo[3,2,1-jk]carbazole and 9H-carbazole building blocks, with high triplet energies (ET > 2.80 eV) and good thermal stability (Tg > 101 °C) were synthesized, characterized and applied as host materials in PhOLED devices. In course of the preparation of the materials, an improved protocol for the synthesis of the indolo[3,2,1-jk]carbazole moiety has been developed. The careful molecular design of the title compounds allowed to avoid excimer formation of the indolo[3,2,1-jk]carbazole subunits in thin films. Therefore, the improved molecular design broadened the applicability of indolo[3,2,1-jk]carbazole host materials and significantly enhanced the efficiency of PhOLED devices based on these derivatives compared to previously reported indolo[3,2,1-jk]carbazole based compounds. Accordingly, employing the newly developed materials red (CEmax: 32.7 cd A−1, PEmax: 31.0 lm W−1, EQEmax: 20.4%), green (CEmax: 81.0 cd A−1, PEmax: 87.4 lm W−1, EQEmax: 21.5%) and blue (CEmax: 35.5 cd A−1, PEmax: 39.9 lm W−1, EQEmax: 18.0%) PhOLED devices with a remarkably low efficiency roll-off at 1000 cd m−2 (Cz2ICz – red: 5%; green: 0%; blue: 6%) were fabricated.
Introduction
Since the first reports on electroluminescence,1 the technology of Organic Light Emitting Diodes (OLEDs) has been steadily developed.2–6 In particular, the introduction of heavy transition metal complexes, which are capable of efficient phosphorescence, boosted the efficiency of OLED devices.7,8 These metal complexes can harvest singlet and triplet excitons simultaneously in phosphorescent OLEDs (PhOLEDs) and thus significantly enhance the internal quantum efficiency compared to traditional purely fluorescent organic materials.9,10 In 2012 Adachi et al. presented a new class of highly efficient emitters based on the concept of thermally activated delayed fluorescence (TADF).11 For this purpose, purely organic donor–acceptor materials with a carefully controlled, small energy splitting between the excited singlet and excited triplet state have been developed. The small energy difference between the two excited states allows for the promotion of the triplet excitons to singlet excitons at room temperature by means of thermal energy. Thus, TADF emitters can match the efficiency of phosphors without the incorporation of heavy metals.12,13Both, phosphorescent and TADF emitters, need to be widely dispersed in a host material to prevent concentration quenching, particularly at a high brightness. Moreover, these host materials provide an enhanced thermal stability and improved charge transport properties of the emitting layer. The triplet energy (ET) of the host material has to be higher than the ET of the emitting material, to confine the excited states on the emitter.14–16
The 9H-carbazole building block has been widely employed in the design of host materials owing to favorable hole transport properties.17–23 In particular, 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) has been frequently employed as host material for various phosphorescent emitters.14 However, CBP features two major drawbacks: (i) a low ET of 2.56 eV,24 which prohibits the application of CBP as host material for blue emitters and (ii) a low glass transition temperature (Tg) of 62 °C.25
Recently, we introduced indolo[3,2,1-jk]carbazole (ICz) as a novel, fully planarized arylamine building block to the field of organic electronics.26,27 The efficient preparation of the ICz motive employing a C–H activation strategy28 enables the application of this structural motive in functional organic materials.26,29 The incorporation of the ICz scaffold into the CBP backbone yielded C2 substituted ICz based host materials (Scheme 1) with high ETs and good thermal stability.30
 | ||
| Scheme 1 Design concept of the novel ICz based host materials. | ||
Furthermore, the ICz motive has been employed as weakly electron accepting unit in the design of efficient blue thermally activated delayed fluorescent (TADF) emitters.31 Substitution of ICz with a cyano group increased the electron accepting properties of the building block and allowed for the preparation of deep blue TADF emitters.32
However, the planar structure of ICz allows for a pronounced, intermolecular interaction between individual host molecules. Consequently, the formation of excimers in thin films of the developed host materials was observed, which prohibited the efficient application of the majority of the developed compounds in blue PhOLEDs.30 The formation of excimers in 9H-carbazole containing materials has been previously described.33–38 However, excimer formation was also observed in ICz based host materials without 9H-carbazole substituents.30 Noteworthy, the energy of the observed excimers was approximately 2.25 eV and thus lower than the energy of the reported 9H-carbazole based excimers (2.4–2.6 eV).34,35,37,38 Hence, it is plausible to assume that the nature of these excimers is different from those found in 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CDBP) and related materials.36,38
Excimers significantly impact the efficiency of blue and, to an even larger extent, deep blue emitters and thus also the practical applicability of OLED devices. Therefore, it is of crucial importance to understand the mechanism behind excimer formation in order to control this undesired interaction. Consequently, the aim of our current work is to improve the molecular design of the ICz based host materials and investigate the effect of the molecular modifications on the excimer formation. The effect of molecular design on the excimer formation in thin films is of particular interest and a deeper understanding of the structural features that lead to excimer formation is crucial to control and avoid this undesired effect. Therefore, compound CzICz (Scheme 1) was revisited, as it exhibited the best performance among the previously reported ICz materials. Notably, the excimers observed for ICz-based host materials were located on the ICz moiety as discussed above. Accordingly, an improved molecular design has to focus on the prevention of excimer formation on this particular molecular building block. The tendency for excimer formation depends strongly on the intermolecular interaction between the individual molecules and their respective alignment.39 Thus, the design concept of the new ICz based materials relies on the prevention of intermolecular interactions of the individual ICz moieties (Scheme 1). To hinder the ordered arrangement of the molecules, we intended to decrease the symmetry of CzICz by altering the substitution position of the attached 9H-carbazole motive from the 2-position to the 5-position of the ICz moiety, thus removing the twofold rotation axis (5CzICz). Furthermore, we introduced additional 9H-carbazole groups in order to increase the steric demand of the substituents and shield the ICz from adjacent molecules (Cz2ICz, Cz3ICz). Consequently, the ICz is continuously moved from the periphery of the molecule to its center.
Results and discussion
Synthesis
Three different approaches were pursued in the preparation of the target materials, owing to the varying substitution positions of the ICz moiety (Scheme 2). Alternative strategies had to be developed to realize the desired substitution patterns, as nucleophilic substitution preferentially occurs in the 2-position of ICz. In the case of 5CzICz, both amine groups were simultaneously introduced by nucleophilic substitution of 1 with 9H-carbazole. Subsequently, C–H activation was utilized to establish the ICz moiety by ring closure. Notably, a novel procedure was employed to accomplish the ring closure. Previously, a preformed Pd-catalyst with an N-heterocyclic carbene ligand40,41 was employed. In the novel protocol, the precursor salt of the ligand (1,3-bis(2,6-diisopropylphenyl)-1H-imidazol-3-ium chloride; Scheme 2) was directly applied in combination with Pd(OAc)2 forming the catalyst in situ. Following this procedure, 5CzICz was obtained in a good yield of 79% after column chromatography. The new procedure is preferred compared to the previously described methodology because it renders the separate formation of the catalyst unnecessary while retaining the high yields of the ring closing step. In analogy, a nucleophilic substitution of 1-bromo-4-fluorobenzene with 2,6-dibromobenzenamine was the first step in the preparation of Cz2ICz. Subsequently, twofold C–H activation established the ICz scaffold. Notably, dinitro-ICz (5) was obtained in an excellent yield of 93%, highlighting the versatility of the novel methodology for the ring closure. Using SnCl2·2H2O, 5 was reduced to diamine 6. Following the Nozaki approach, the two 9H-carbazole moieties were formed by Pd catalyzed Buchwald–Hartwig amination, yielding Cz2ICz. The preparation of Cz3ICz was accomplished by threefold bromination of ICz followed by the introduction of the 9H-carbazoles in an Ullmann reaction. All target materials were characterized by 1H and 13C NMR as well as high resolution mass spectrometry.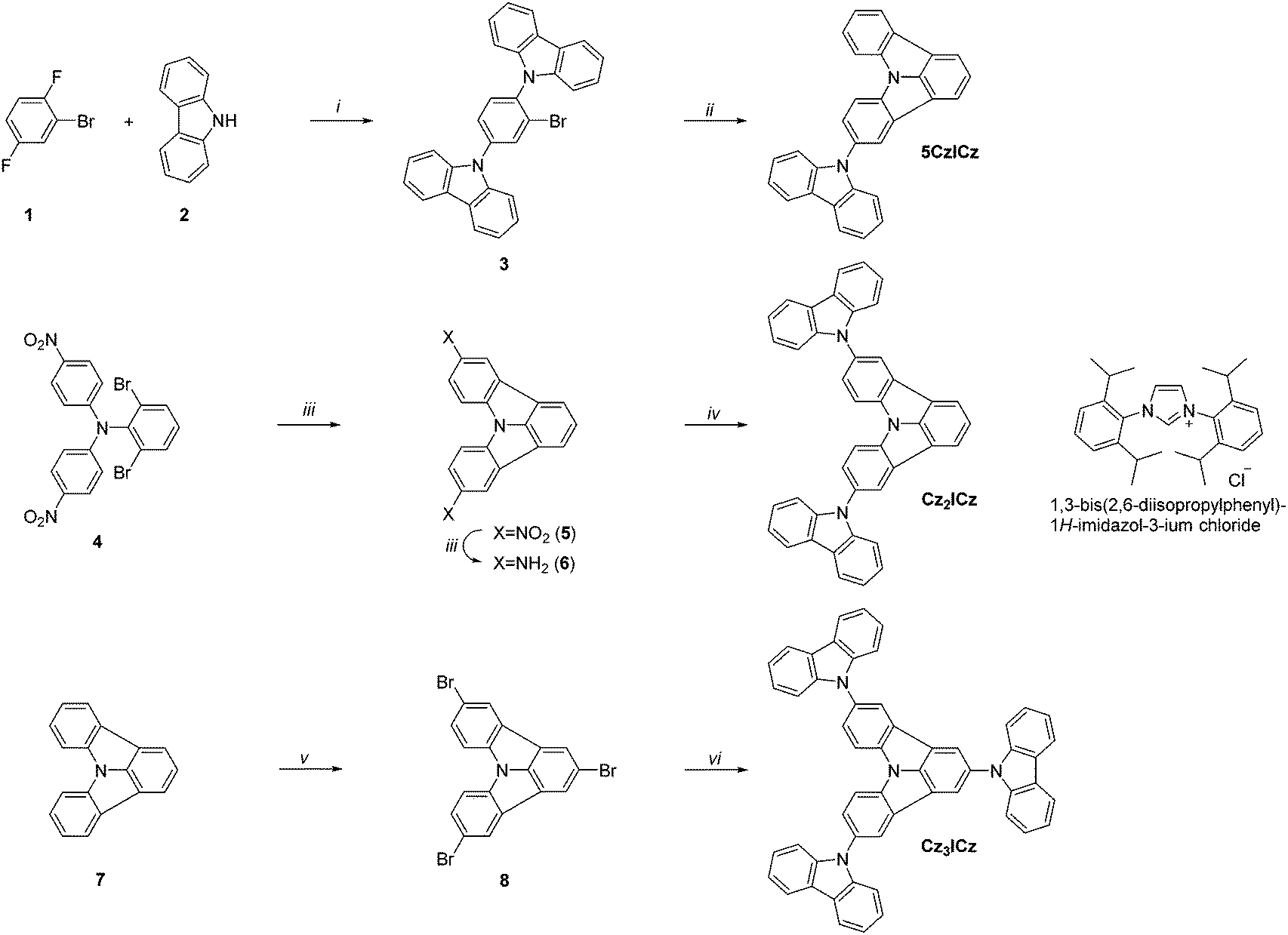 | ||
Scheme 2 Synthetic approach towards the ICz based host materials: (i) Cs2CO3, DMSO, 120 °C; (ii) Pd(OAc)2, ligand·HCl, K2CO3, DMA, 130 °C; (iii) SnCl2·2H2O, DMSO/H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 80 °C; (iv) NaOtBu, 2-bromo-2′-iodo-1,1′-biphenyl, Pd2(dba)3, dppf, toluene, reflux; (v) Br2, DCM (dichloromethane); (vi) 9H-carbazole, CuSO4·5H2O, K2CO3, 250 °C. :1), 80 °C; (iv) NaOtBu, 2-bromo-2′-iodo-1,1′-biphenyl, Pd2(dba)3, dppf, toluene, reflux; (v) Br2, DCM (dichloromethane); (vi) 9H-carbazole, CuSO4·5H2O, K2CO3, 250 °C. | ||
In the case of 5CzICz, the molecular structure was determined by single crystal analysis (Fig. 1). The ICz and 9H-carbazole moieties are virtually flat [maximum distance to least squares (LS) planes defined by the non-H atoms: 0.043(2) Å (ICz) and 0.029(4) Å (9H-carbazole)] and distinctly inclined to each other [angle between LS planes: 66.01(6)°]. Notably, the substitution pattern of the ICz influences the torsion angle between the two planar subunits. Whereas the torsion angle in C2 substituted CzICz (Scheme 1) was 54.07(5)°,30 it is increased in C5 substituted 5CzICz.
 | ||
| Fig. 1 Molecular structure of 5CzICz; C and N atoms are represented by grey and blue ellipsoids drawn at 50% probability level, H atoms by white spheres of arbitrary radius. | ||
The thermal properties of the developed host materials were investigated by differential scanning calorimetry (DSC, Fig. S16, ESI†) and thermogravimetric analysis (TGA, Fig. S17, ESI†). The Tg of the materials increased with progressive 9H-carbazole substitution from 5CzICz (101 °C) to Cz2ICz (171 °C) and Cz3ICz (327 °C). The Tg of all compounds is significantly higher compared to CBP,25 which can be attributed to the increased rigidity of the ICz scaffold and benefits the morphological stability of thin films of the materials. Furthermore, all materials exhibited an exceptional thermal stability with decomposition temperatures (Td: corresponding to 5% mass loss during thermogravimetric analysis) of 450 °C or higher.
The energies of the HOMO and LUMO levels of the newly developed ICz based host materials were determined by cyclic voltammetry (CV, Fig. S18–S20, ESI†). All compounds exhibited irreversible oxidation, as typically found for 9H-carbazole and ICz derivatives, due to the instability of the formed oxidation products.30,42 The energy of the HOMO levels span a narrow range between −5.57 and −5.63 eV, comparable to CzICz.30 The LUMO levels of 5CzICz and Cz2ICz are located at −2.47 eV, while the LUMO of Cz3ICz is located slightly lower at −2.59 eV. These HOMO and LUMO energy levels indicate no significant injection barriers for holes and electrons from adjacent layers in OLED devices.
Theoretical calculations
To obtain a more detailed understanding of the molecular electronics, we employed density functional theory (DFT). The calculated energy of HOMO levels are in good agreement with the experimentally determined values with a maximum deviation of 0.04 eV (Table 1). In contrast to the HOMO levels the energy of LUMO levels are systematically overestimated. Furthermore, the theoretical calculations predict a continuous decrease of the LUMO energy from 5CzICz to Cz2ICz and Cz3ICz, which is only observed to a small degree for Cz3ICz in the experimental data. A clear separation of the frontier molecular orbitals is observed for all three materials (Fig. 2). The HOMOs are mainly located on the 9H-carbazole units with some contribution from the central nitrogen of the ICz scaffold. This behavior is in line with the stronger electron donating nature of the 9H-carbazole compared to ICz.26 Likewise, the LUMO levels of all materials are exclusively located on the ICz moieties.| T g/Trc/Tm/Tda [°C] | Opt. BGb,c [eV] | λ max,PL [nm] | HOMO/LUMO [eV] | E T [eV] | ||
|---|---|---|---|---|---|---|
| Exp.d | Cal.e | |||||
| a Determined by thermogravimetric analysis and differential scanning calorimetry; Tg: glass transition temperature; Trc: recrystallization temperature; Tm: melting point; Td: decomposition temperature corresponding to 5% mass loss. b Optical bandgap determined from the absorption onset. c Measured in DCM solutions (5 μM) at room temperature. d Calculated from the onset of the oxidation and reduction peak observed during cyclic voltammetry. e Calculated applying density functional theory level (B3LYP/6-311G**). f Determined from the highest vibronic transition in solid solutions of a mixture of DCM/toluene/MeOH (10/10/1) at 77 K. g Not observed. | ||||||
| 5CzICz | 101/181/244/450 | 3.19 | 410 | −5.57/−2.47 | −5.53/−1.77 | 2.83 |
| Cz2ICz | 171/n.o.g/308/>500 | 3.08 | 420 | −5.54/−2.47 | −5.57/−1.96 | 2.82 |
| Cz3ICz | 327/n.o.g/n.o.g/>500 | 3.02 | 432 | −5.63/−2.59 | −5.62/−2.17 | 2.80 |
 | ||
| Fig. 2 Spatial distribution of the HOMOs and LUMOs of the new host materials. | ||
To learn more about the nature of the excited states, we used time dependent density functional theory (TD-DFT) to calculate the S0 → S1 and S0 → T1 transitions. The natural transition orbitals (NTOs) of the first excited singlet state of all three materials closely resemble the HOMOs and LUMOs of the compounds (Fig. S15, ESI†). Accordingly, the excited holes are localized on the 9H-carbazole units, whereas the excited electrons are localized on the ICz motive. Therefore, the S1 is an intramolecular charge transfer (ICT) state. In contrast, for the S0 → T1 transition the excited hole and the excited electron are both localized on the ICz moiety with a small contribution to the excited holes coming from the nitrogen atoms of the 9H-carbazole units and to some extent also from the 9H-carbazoles in the case of Cz3ICz (Fig. 3). Thus, the T1 state can be considered a mainly localized excited (LE) state centered on the ICz with a light charge transfer character in the case of Cz2ICz and Cz3ICz.
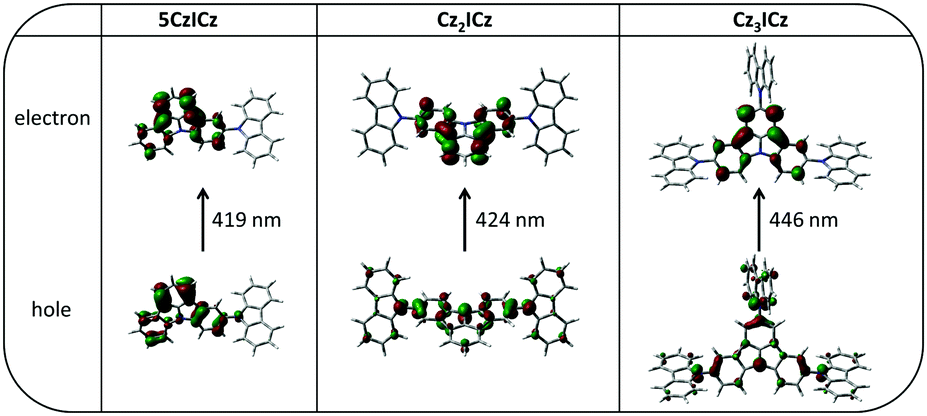 | ||
| Fig. 3 Natural transition orbitals (NTOs) of the S0 → T1 transition calculated by TD-DFT at the optimized S0 geometries. | ||
Photophysical properties
UV/vis absorption, room temperature photoluminescence and low temperature phosphorescence spectra were recorded to investigate the photophysical properties of the newly developed materials (Fig. 4). All three materials featured two intense absorption bands between 286 and 297 nm, which can be attributed to π–π* transitions centered on the 9H-carbazole and ICz moieties, respectively. Moreover, two transitions of the conjugated backbone of the molecules are observed between 327 and 330 nm and between 341 and 344 nm as small peaks or shoulders for all three molecules. Notably, the absorption wavelength of the lowest energy transition is continuously red-shifted from 5CzICz (365 nm) to Cz2ICz (369 nm) and Cz3ICz (389 nm), owing to the increased size of the π-conjugated system. Accordingly, the absorption onsets are shifted towards lower energy from 3.19 (5CzICz) to 3.08 (Cz2ICz) and 2.97 eV (Cz3ICz). Compared to CBP, the optical bandgaps are significantly reduced, which is expected to decrease the driving voltage and thus yield high power efficiencies in PhOLED devices.30 | ||
| Fig. 4 UV/vis absorption (dashed lines), normalized fluorescence (solid lines) and normalized phosphorescence (dotted lines) spectra of the ICz based host materials. UV/vis absorption and normalized fluorescence spectra were recorded in DCM solutions (5 μM) at room temperature. Phosphorescence spectra were recorded in solid solutions of DCM/toluene/MeOH (10/10/1) at 77 K. F = fluorescence; P = phosphorescence. | ||
All materials exhibited unstructured fluorescent emission in DCM (dichloromethane) at room temperature. This observation is in good agreement with the theoretical calculations, which suggest an ICT S1 state. Furthermore, the absorption and emission properties of the materials in solvents with different polarity were investigated (Fig. S24–S26, ESI†). While the absorption was basically independent of the solvent polarity, all materials exhibited a distinct solvatochromism and the emission maxima were shifted towards higher wavelengths with increasing solvent polarity. This behavior indicates a polar excited state and thus confirms the assumption of an ICT S1 state.
In analogy to the absorption onsets, the wavelengths of the fluorescence maxima are red-shifted from 410 (5CzICz) to 420 (Cz2ICz) and 432 nm (Cz3ICz). In contrast to the slightly different fluorescence, 5CzICz and Cz2ICz featured nearly the same vibronically resolved phosphorescent emission. The maxima of the highest energy vibronic transitions are located at 438 and 440 nm, corresponding to ETs of 2.83 and 2.82 eV, respectively. The presence of a vibronically resolved emission confirms the LE nature of the T1 state, as suggested by the theoretical calculations. The phosphorescence spectrum of Cz3ICz appears less structured compared to the other two materials, due to the increased CT character. Nonetheless, the same transitions can be found with a slight overall red-shift leading to an ET of 2.80 eV. From these findings it can be concluded that the nature of the excited triplet state of the three materials is indeed basically the same and localized on the ICz moiety as predicted by the theoretical calculations.
Notably, the ETs of the compounds are significantly increased compared to CBP24 and in the same range as those of previously reported ICz derivatives.30 Therefore, the developed materials are potential host materials for blue phosphorescent emitters.
To understand the effects of the molecular modifications on excimer formation, the photoluminescence of thin films of the newly designed ICz based host materials were recorded. Compared to the emission from diluted DCM solutions, the film fluorescence of the compounds is slightly red-shifted (Fig. 5), owing to the altered chemical environment of the individual molecules. Cz3ICz exhibited a rather sharp emission band. This behaviour may be attributed to the fact that the excited state is most shielded by the threefold carbazole substitution (Fig. S15, ESI†) of Cz3ICz and thus less influenced by the chemical environment. Most notably, there are no additional low energy emission bands in the emission of the thin films, as previously observed for the C2 substituted ICz based host materials.30 The absence of this long wavelength emission, which is a sign for the formation of excimers,34,36 proves that the presented design approach of the novel materials indeed sufficiently suppresses the intermolecular interaction of the individual ICz units and therefore prevents the formation of excimer states in thin films.
 | ||
| Fig. 5 Normalized fluorescence and spectra of diluted DCM solutions and thin films of the novel ICz based host materials. | ||
Electroluminescent properties
Finally, the developed compounds were investigated regarding their universal applicability as host materials in red, green, and blue PhOLEDs. Accordingly, employing 5CzICz (1), Cz2ICz (2), and Cz3ICz (3) as host materials red (R), green (G), and blue (B) PhOLED prototype devices with the following architecture have been fabricated: (i) R: ITO/MoO3 (8 nm)/TAPC (50 nm)/EML (host material, Ir(MDQ)2(acac) (5%), 12 nm)/BPhen (70 nm)/LiF/Al; (ii) G: ITO/MoO3 (8 nm)/TAPC (60 nm)/EML (host material, Ir(ppy)2(acac) (8%), 12 nm)/BPhen (70 nm)/LiF/Al; (iii) B: ITO/MoO3 (8 nm)/TAPC (50 nm)/EML (host material, FIrpic (10%), 15 nm)/BmPyPB (45 nm)/LiF/Al. TAPC was utilized as a hole transporting layer and BPhen was employed as an electron transporting and hole blocking layer. In the case of blue devices, BPhen was replaced by BmPyPB, owing to the ET of BmPYPB (2.69 eV)43 being higher compared to the ET of BPhen (2.56 eV).44 Current density–voltage–luminance and current efficiency–luminance–power efficiency curves are depicted in Fig. 6. The key electroluminescent properties of all devices are summarized in Table 2. The observed emission exclusively originated from the employed phosphorescent emitter and representative emission spectra of the PhOLED devices are given in the ESI.†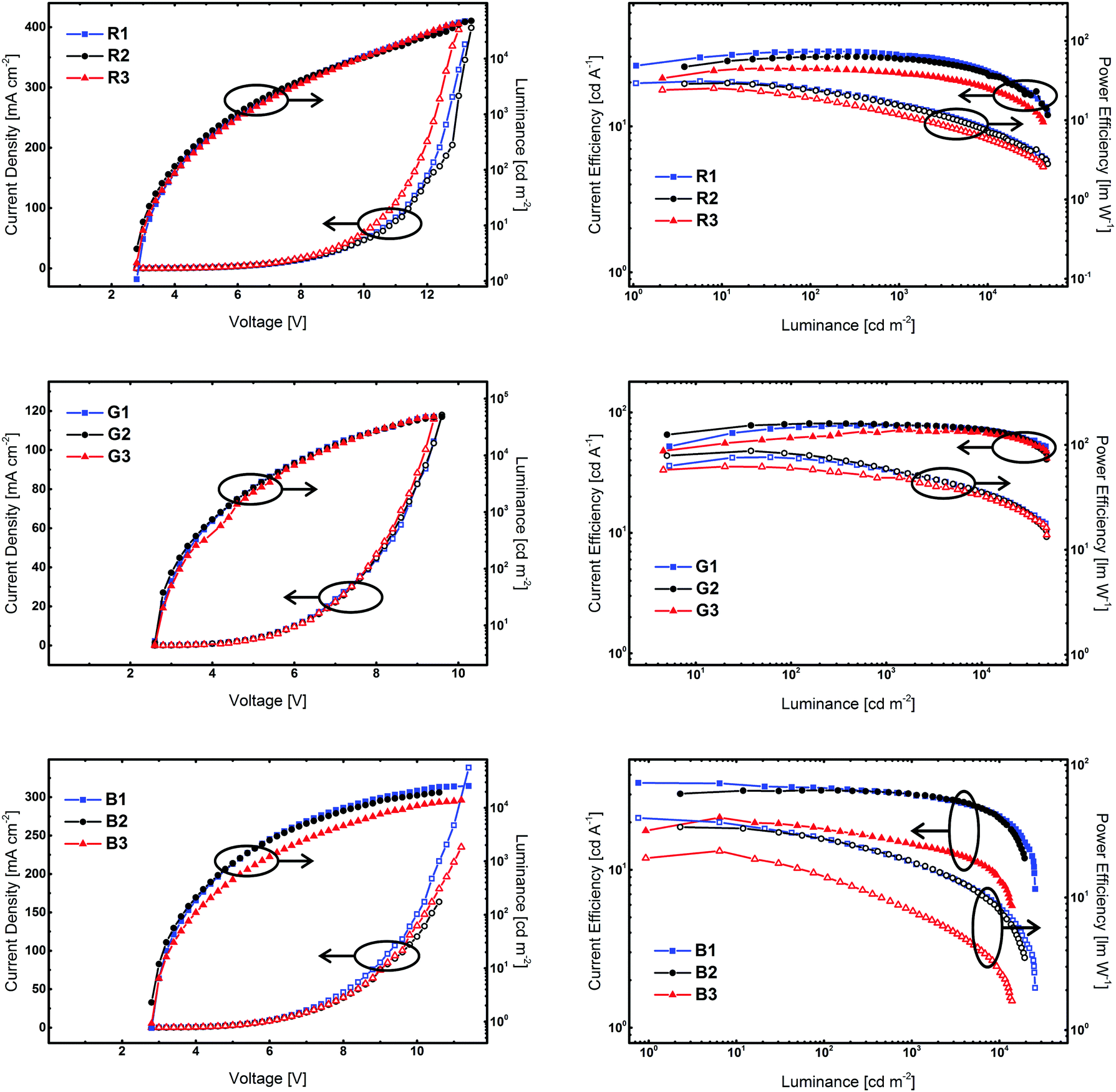 | ||
| Fig. 6 Key electroluminescent properties of the prototype devices employing 5CzICz, Cz2ICz, and Cz3ICz as host materials. Left: Current density–voltage–luminance (current density: hollow symbols; luminance: full symbols), right: current efficiency–luminance–power efficiency (current efficiency: full symbols; power efficiency: hollow symbols). | ||
| V on [V] | CEa [cd A−1] | PEa [lm W−1] | EQEa [%] | |
|---|---|---|---|---|
| a Measured at a brightness of 1000 cd m−2/measured at a brightness of 5000 cd m−2/maximum efficiency. | ||||
| R1 | 2.8 | 30.7/27.0/32.7 | 15.8/10.1/31.0 | 18.7/16.1/20.2 |
| R2 | 2.8 | 29.0/25.9/30.0 | 15.4/9.7/29.3 | 19.3/17.0/20.4 |
| R3 | 2.8 | 22.9/20.4/24.9 | 11.5/7.4/25.2 | 16.3/14.2/17.6 |
| G1 | 2.6 | 77.7/75.1/78.0 | 57.1/42.1/76.3 | 20.7/20.1/20.8 |
| G2 | 2.6 | 79.3/75.6/81.0 | 59.1/42.5/87.4 | 21.0/20.1/21.5 |
| G3 | 2.6 | 69.2/69.7/71.8 | 48.5/38.8/62.2 | 18.6/18.6/19.2 |
| B1 | 2.8 | 29.8/25.1/35.5 | 18.3/11.5/39.9 | 15.0/12.7/18.0 |
| B2 | 2.8 | 30.1/25.8/31.7 | 18.5/11.7/34.0 | 14.6/12.5/15.4 |
| B3 | 2.8 | 14.6/11.5/21.4 | 7.9/4.4/22.5 | 7.0/5.5/10.2 |
Notably, all devices featured a very low onset voltage (Von) of 2.6 V (G1–3) or 2.8 V (R1–3 and B1–3). The Von is thus slightly lower than the electrochemically determined HOMO–LUMO gap and indicates efficient charge injection into the emitting layer. The slightly higher Von of the red devices compared to the green devices is attributed to direct charge trapping on the red emitter.
Red devices R1–3 featured a good performance with a maximum external quantum efficiency (EQEmax) of up to 20.4% for R2. Among these devices, R1 employing 5CzICz as host material featured the highest maximum current efficiency (CEmax) of 32.7 cd A−1 and the highest maximum power efficiency (PEmax) of 31.0 lm W−1. Notably, the CE of R1 decreased only slightly with increasing brightness and the CEs at 1000 and 5000 cd m−2 were 30.7 and 27.0 cd A−1, corresponding to an efficiency roll-off of 6% and 17%, respectively. A good efficiency at high brightness is of particular importance for practical applications and thus is a relevant characteristic of newly developed host materials. The CEmax and PEmax of R2 and R3 of 30.0 and 24.9 cd A−1 and 29.3 and 25.2 lm W−1 were slightly lower compared to the CEmax and PEmax of R1. The efficiency roll-off of R2 and R3, however, was similar compared to R1.
In analogy to R1–3, green devices G1–3 exhibited good performance with very high CEmax of 81.0 cd A−1 for G2, 78.0 cd A−1 for G1, and 71.8 cd A−1 for G3, corresponding to EQEmax of 21.5%, 20.8%, and 19.2%. Owing to the low driving voltages, the devices also displayed high PEmax of 76.3 (G1), 87.4 (G2), and 62.2 lm W−1 (G3). Most strikingly however, G1 showed hardly any efficiency roll-off at 1000 cd m−2. It still exhibited an excellent CE of 75.1 cd A−1 and a PE of 42.1 lm W−1 at 5000 cd m−2 corresponding to an EQE of 20.1% and an efficiency roll-off of 4%. Likewise, the efficiency roll-off in G2 was low. At 5000 cd m−2G2 displayed a CE of 75.6 cd A−1 and an EQE of 20.1% and thus an efficiency roll-off of 7%. In the case of device G3, the situation is slightly different as the efficiency of G3 increased at lower and intermediate brightness and its CEmax was reached at approximately 1700 cd m−2, before the CE slowly decreased. As can be seen in Fig. 6 (right column, middle row), the CE and PE of G3 are slightly lower compared to G1 and G2 at lower brightness but approach those of the other two devices at higher brightness. Therefore, G3 exhibited the lowest efficiency roll-off of only 3% at 5000 cd m−2 among all green devices.
Notably, the performance of the green devices employing the newly developed host materials could be significantly improved compared to our previous results with C2 substituted ICz derivatives.30 While the EQEs were increased from 15 to 20% the PE of the devices employing the new materials were doubled over the whole brightness range highlighting the effect of the improved molecular design of the title compounds. The high CE and EQE of the red and green devices indicate efficient exciton confinement on the employed phosphorescent emitters. Moreover, the high power efficiencies result from the low bandgap of the materials and efficient charge injection into the emitting layer. These device characteristics are vital for the practical applications and underline the importance of the ICz based architecture as a novel platform for the development of efficient host materials.
While the three developed materials featured comparable results for red and green emitters, the situation is different for blue emitting FIrpic. Device B1 employing 5CzICz exhibited a very satisfying CEmax of 35.5 cd A−1 corresponding to an EQEmax of 18.0% and a high PEmax of 39.9 lm W−1, while the CEmax (31.7 cd A−1) and EQEmax (15.4%) of B2 were somewhat lower. At higher brightness, B1 and B2 featured similar efficiency values. On the other hand, the performance of Cz3ICz in B3 was considerably lower with an EQEmax of 10.2%. Moreover, B3 displayed the highest efficiency roll-off among the investigated materials. In summary, the observed efficiencies of devices B1 and B2 are very satisfying. In analogy to the green devices, the novel design concept proved superior to the C2 substitution pattern. Not only were all new materials applicable as host materials for FIrpic, but also B1 and B2 exhibited increased device performance compared to our previous results.30
Conclusions
In this study, we report on three novel indolo[3,2,1-jk]carbazole based host materials for PhOLEDs. Our systematic investigations regarding the substitution position of the indolo[3,2,1-jk]carbazole moiety revealed that the formation of excimers on the indolo[3,2,1-jk]carbazole subunits in thin films of the materials can be prevented by a careful molecular design. Clearly, the suggested design strategy for indolo[3,2,1-jk]carbazole based host materials starting with C5 substitution proved to be superior compared to C2 substitution. Accordingly, the efficiency of PhOLED devices based on the developed materials could be improved compared to previously reported derivatives and all new materials were applicable as host materials in blue devices. Furthermore, the efficiency roll-off of the fabricated prototype devices was remarkably low. Therefore, we are convinced that these investigation will guide the design of new indolo[3,2,1-jk]carbazole derivatives for applications as host materials and beyond.Experimental section
General information
All reagents and solvents were obtained commercially and used without further purification. Anhydrous solvents were prepared by filtration through drying columns. The water content of purchased DMA was determined by Karl Fischer titration and corrected to 1000 ppm for C–H activation reactions. Column chromatography was performed using silica 60 (Merck, 40–63 μm). NMR spectra were recorded on a Bruker DRX-600 MHz Spectrometer. Absorption and photoluminescence measurements were conducted using a Thermo Scientific NanoDrop OneC Microvolume UV/vis spectrophotometer and a PerkinElmer LS 55 fluorescence spectrometer, respectively. Solution measurements were recorded employing DCM solutions (5 μM). Phosphorescence spectra were recorded at 77 K using solid solutions of the materials in a mixture of DCM/toluene/MeOH (10/10/1) with a delay of 0.1 ms or thin films of the compounds and a delay of 1 ms. Thermal analysis was carried out with a heating rate of 10 K min−1. Differential scanning calorimetry (DSC) was performed using a Netzsch DSC 200 F3 Maia and a Netzsch simultaneous thermal analyzer (STA 449 F1 Jupiter) was employed for thermogravimetric analysis (TGA), working with pierced aluminum pans and under N2 (TGA) or argon (DSC) atmosphere. Cyclic voltammetry (CV) was done using a three electrode configuration consisting of a Pt working electrode, a Pt counter electrode, and an Ag/AgCl reference electrode and a PGSTAT128N potentiostat provided by Metrohm Autolab B.V. Measurements were carried out in a 0.5 mM solution in anhydrous DCM (oxidation) or ACN (reduction) employing Bu4NBF4 (0.1 M) as supporting electrolyte. Prior to the measurements, the solutions were purged with nitrogen for approximately 15 minutes. The HOMO and LUMO energy levels were calculated from the onset of the oxidation and reduction peaks, respectively. The onset potential was determined by the intersection of two tangents drawn at the background and the rising of the oxidation and reduction peaks. High resolution mass spectroscopy was carried out using an Agilent 1100/1200 HPLC in combination with an Agilent 5230 AJS ESI-TOF mass spectrometer.Synthetic details
1, 2, and 9H-carbazole have been purchased from commercial suppliers and used without further purification. Indolo[3,2,1-jk]carbazole26 and 2-bromo-2′-iodo-1,1′-biphenyl45 have been prepared according to published procedures.:DCM 85:15 → 83:17). 1H-NMR (600 MHz, CD2Cl2): δ = 8.22–8.20 (m, 4H), 8.16 (d, J = 2.3 Hz, 1H), 7.83 (dd, J = 8.3, 2.3 Hz, 1H), 7.77 (d, J = 8.2 Hz, 1H), 7.65 (d, J = 8.2 Hz, 2H), 7.53–7.48 (m, 4H), 7.39–7.34 (m, 4H), 7.26 (d, J = 8.2 Hz, 2H) ppm. 13C NMR (150 MHz, CD2Cl2): δ = 141.4, 140.9, 139.7, 135.9, 132.7, 132.7, 127.8, 126.9, 126.7, 125.1, 124.3, 123.8, 121.2, 121.0, 120.9, 120.8, 110.6, 110.2 ppm. HRMS (ESI): m/z calculated for C30H19BrN2: 486.0732 [M]+; found: 486.0723 [M]+.
:DCM 85:15). Single crystals of 5CzICz were obtained after crystallization from toluene. 1H-NMR (600 MHz, CDCl3): δ = 8.31 (d, J = 1.9 Hz, 1H), 8.22–8.19 (m, 3H), 8.12–8.11 (m, 2H), 8.04 (d, J = 7.4 Hz, 1H), 7.99 (d, J = 7.9 Hz, 1H), 7.74 (dd, J = 8.3, 2.0 Hz, 1H), 7.64–7.61 (m, 2H), 7.47–7.42 (m, 5H), 7.35–7.32 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3): δ = 144.4, 141.6, 138.6, 137.6, 131.5, 131.3, 130.0, 127.0, 126.0, 125.9, 123.3, 123.2, 122.4, 122.1, 120.3, 120.1, 119.8, 119.8, 118.8, 118.0, 112.9, 112.2, 109.7 ppm (one carbon atom was not detected). HRMS (ESI): m/z calculated for C30H18Br2: 406.1470 [M]+; found: 406.1468 [M]+.
:1) in a three necked flask under argon atmosphere and heated to 80 °C overnight. After being cooled to room temperature, the reaction mixture was basified with 2 M NaOH and repeatedly extracted with CHCl3. The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. Column chromatography (light petrol:EA 1:3 → EA) yielded 6 (0.64 g, 2.4 mmol, 59%) as a yellow-green solid. 1H-NMR (600 MHz, CD2Cl2): δ = 7.96 (d, J = 7.3 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 7.49 (t, J = 7.4 Hz, 1H), 7.47 (d, J = 2.3 Hz, 2H), 6.93 (dd, J = 8.5, 2.3 Hz, 2H), 3.78 (s, 4H) ppm. 13C NMR (150 MHz, CD2Cl2): δ = 145.0, 142.0, 133.1, 131.0, 122.2, 119.7, 118.8, 115.2, 112.6, 109.7 ppm. HRMS (ESI): m/z calculated for C18H13N3: 271.1109 [M]+; found: 271.1109 [M]+.
:DCM 85:15 → 33:67). 1H-NMR (600 MHz, CDCl3): δ = 8.37 (d, J = 1.9 Hz, 2H), 8.22 (d, J = 7.8 Hz, 4H), 8.20 (d, J = 8.3 Hz, 2H), 8.12 (d, J = 7.4 Hz, 2H), 7.80 (dd, J = 8.3, 2.1 Hz, 2H), 7.67 (t, J = 7.4 Hz, 1H), 7.49–7.45 (m, 8H), 7.36–7.33 (m, 4H) ppm. 13C NMR (150 MHz, CDCl3): δ = 145.1, 141.5, 137.6, 131.9, 131.4, 126.3, 126.0, 123.6, 123.3, 122.6, 120.5, 120.4, 119.9, 118.4, 113.0, 109.7 ppm. HRMS (ESI): m/z calculated for C42H25N3: 571.2048 [M]+; found: 571.2053 [M]+.
:DCM 70:30) yielded Cz3ICz (2.25 g, 3.1 mmol, 76%) as beige solid. 1H-NMR (600 MHz, CDCl3): δ = 8.34 (d, J = 2.0 Hz, 2H), 8.24–8.23 (m, 4H), 8.21–8.19 (m, 6H), 7.86 (dd, J = 8.5, 2.1 Hz, 2H), 7.50–7.45 (m, 8H), 7.43–7.41 (m, 2H), 7.38 (d, J = 8.5 Hz, 2H), 7.35–7.30 (m, 6H). 13C NMR (150 MHz, CDCl3): δ = 144.0, 142.2, 141.4, 138.1, 133.6, 132.4, 131.1, 127.0, 126.0, 126.0, 123.3, 123.1, 122.9, 120.9, 120.4, 120.3, 120.0, 119.9, 119.1, 113.3, 109.6, 109.6 ppm. HRMS (ESI): m/z calculated for C54H32N4: 736.2627 [M]+; found: 736.2624 [M]+.
Computational details
(TD-)DFT calculations were performed using the Gaussian 09 package49 applying the Becke three-parameter hybrid functional with Lee–Yang–Perdew correlation (B3LYP)50,51 in combination with Pople basis sets 6-311G(d,p) or 6-311+G(d,p).52 Geometry optimizations were performed in the gas phase and without symmetry constraints applying the 6-311G(d,p) basis sets. Singlet and triplet excitations were calculated using the S0 optimized geometry applying the 6-311+G(d,p) basis sets. Orbital plots were generated using GaussView.53Single crystal diffraction
Intensity data of 5CzICz were collected at 100 K in a dry stream of nitrogen using Cu Kα radiation generated by microfocus tube on a Rigaku Oxford Diffraction XtaLAB Synergy diffractometer equipped with a Pilatus 200 K detector. Data reduction and absorption correction were performed with the CrysAlisPro software suite.54 The structure was solved with the dual-space method implemented in SHELXT55 and refined against F2 with SHELXL.56 H atoms were placed at calculated positions and refined as riding on the parent C atom.Device fabrication and measurement
Prior to device fabrication, the spectroscopically pure host materials were purified by crystallization from toluene (5CzICz, Cz2ICz, Cz3ICz) followed by sublimation (5CzICz, Cz2ICz). The sheet resistance of the patterned ITO-coated glass substrates used was 10 Ω per square. Before vacuum evaporation, they were completely cleaned in the ultrasonic bath using detergents and deionized water in turn. After that, the substrates were put into a vacuum drying oven at a temperature of 120 °C for 15 min. Then, they were treated with UV generated ozone for 15 min. All the organic layers were grown by thermal evaporation under high vacuum of ∼10−4 Pa. The evaporation rate of organic layers was 1–2 Å s−1 Current–voltage–brightness characteristics and EL spectra were carried out by a Keithley source measurement unit (Keithley 2400 and Keithley 2000) with a calibrated silicon photodiode and Spectrascan PR650 spectrophotometer, respectively. EQEs were obtained from the luminance, current density, and the EL spectra.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. K. and J. F. gratefully acknowledge financial support by the Austrian Science Fund (FWF) (grant no. I 2589-N34). J. C. and D. M. gratefully acknowledge financial support by the National Natural Science Foundation of China (grant no. 11661131001).Notes and references
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef PubMed.
- Y. Sun, N. C. Giebink, H. Kanno, B. Ma, M. E. Thompson and S. R. Forrest, Nature, 2006, 440, 908–912 CrossRef PubMed.
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lussem and K. Leo, Nature, 2009, 459, 234–238 CrossRef PubMed.
- M. C. Gather, A. Köhnen and K. Meerholz, Adv. Mater., 2011, 23, 233–248 CrossRef PubMed.
- S. Reineke, M. Thomschke, B. Lüssem and K. Leo, Rev. Mod. Phys., 2013, 85, 1245–1293 CrossRef.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4–6 CrossRef.
- M. A. Baldo, D. F. O’Brien, M. E. Thompson and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 14422–14428 CrossRef.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048–5051 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. H. Li, R. F. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC.
- A. Chaskar, H.-F. Chen and K.-T. Wong, Adv. Mater., 2011, 23, 3876–3895 CrossRef PubMed.
- K. S. Yook and J. Y. Lee, Adv. Mater., 2012, 24, 3169–3190 CrossRef PubMed.
- R. J. Holmes, S. R. Forrest, Y.-J. Tung, R. C. Kwong, J. J. Brown, S. Garon and M. E. Thompson, Appl. Phys. Lett., 2003, 82, 2422–2424 CrossRef.
- C. H. Chen, W. S. Huang, M. Y. Lai, W. C. Tsao, J. T. Lin, Y. H. Wu, T. H. Ke, L. Y. Chen and C. C. Wu, Adv. Funct. Mater., 2009, 19, 2661–2670 CrossRef.
- Y. Kuwabara, H. Ogawa, H. Inada, N. Noma and Y. Shirota, Adv. Mater., 1994, 6, 677–679 CrossRef.
- Z. Ge, T. Hayakawa, S. Ando, M. Ueda, T. Akiike, H. Miyamoto, T. Kajita and M.-a. Kakimoto, Org. Lett., 2008, 10, 421–424 CrossRef PubMed.
- S.-J. Su, H. Sasabe, T. Takeda and J. Kido, Chem. Mater., 2008, 20, 1691–1693 CrossRef.
- P. Kautny, Z. Wu, B. Stöger, A. Tissot, E. Horkel, J. Chen, D. Ma, H. Hagemann, J. Fröhlich and D. Lumpi, Org. Electron., 2015, 17, 216–228 CrossRef.
- Y. Tao, Q. Wang, C. Yang, C. Zhong, K. Zhang, J. Qin and D. Ma, Adv. Funct. Mater., 2010, 20, 304–311 CrossRef.
- C. Adachi, R. C. Kwong, P. Djurovich, V. Adamovich, M. A. Baldo, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 2001, 79, 2082–2084 CrossRef.
- M. H. Tsai, Y. H. Hong, C. H. Chang, H. C. Su, C. C. Wu, A. Matoliukstyte, J. Simokaitiene, S. Grigalevicius, J. V. Grazulevicius and C. P. Hsu, Adv. Mater., 2007, 19, 862–866 CrossRef.
- P. Kautny, D. Lumpi, Y. Wang, A. Tissot, J. Bintinger, E. Horkel, B. Stöger, C. Hametner, H. Hagemann, D. Ma and J. Fröhlich, J. Mater. Chem. C, 2014, 2, 2069–2081 RSC.
- H. Puntscher, P. Kautny, B. Stoger, A. Tissot, C. Hametner, H. R. Hagemann, J. Frohlich, T. Baumgartner and D. Lumpi, RSC Adv., 2015, 5, 93797–93807 RSC.
- L.-C. Campeau, P. Thansandote and K. Fagnou, Org. Lett., 2005, 7, 1857–1860 CrossRef PubMed.
- J. Lv, Q. Liu, J. Tang, F. Perdih and K. Kranjc, Tetrahedron Lett., 2012, 53, 5248–5252 CrossRef.
- P. Kautny, Z. Wu, J. Eichelter, E. Horkel, B. Stöger, J. Chen, D. Ma, J. Fröhlich and D. Lumpi, Org. Electron., 2016, 34, 237–245 CrossRef.
- J.-A. Seo, Y. Im, S. H. Han, C. W. Lee and J. Y. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 37864–37872 CrossRef PubMed.
- Y. Im and J. Y. Lee, Chem. Commun., 2013, 49, 5948–5950 RSC.
- W. Klöpffer and D. Fischer, J. Polym. Sci., Polym. Symp., 1973, 40, 43–56 CrossRef.
- K. Tani, Y. Tohda, H. Takemura, H. Ohkita, S. Ito and M. Yamamoto, Chem. Commun., 2001, 1914–1915 RSC.
- V. Jankus and A. P. Monkman, Adv. Funct. Mater., 2011, 21, 3350–3356 CrossRef.
- S. T. Hoffmann, P. Schrögel, M. Rothmann, R. Q. Albuquerque, P. Strohriegl and A. Köhler, J. Phys. Chem. B, 2011, 115, 414–421 CrossRef PubMed.
- S. A. Bagnich, S. Athanasopoulos, A. Rudnick, P. Schrögel, I. Bauer, N. C. Greenham, P. Strohriegl and A. Köhler, J. Phys. Chem. C, 2015, 119, 2380–2387 CrossRef.
- S. A. Bagnich, A. Rudnick, P. Schrögel, P. Strohriegl and A. Köhler, Philos. Trans. R. Soc., A, 2015, 373, 20140446 CrossRef PubMed.
- K. L. Woon, Z. A. Hasan, B. K. Ong, A. Ariffin, R. Griniene, S. Grigalevicius and S.-A. Chen, RSC Adv., 2015, 5, 59960–59969 RSC.
- O. Navarro and S. P. Nolan, Synthesis, 2006, 366–367 Search PubMed.
- N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef PubMed.
- E. Mondal, W.-Y. Hung, H.-C. Dai and K.-T. Wong, Adv. Funct. Mater., 2013, 23, 3096–3105 CrossRef.
- S.-J. Su, E. Gonmori, H. Sasabe and J. Kido, Adv. Mater., 2008, 20, 4189–4194 Search PubMed.
- L. Zhu, K. Xu, Y. Wang, J. Chen and D. Ma, Front. Optoelectron., 2015, 8, 439–444 CrossRef.
- F. Leroux and M. Schlosser, Angew. Chem., Int. Ed., 2002, 41, 4272–4274 CrossRef PubMed.
- P. Kautny, J. Fröhlich, B. Stöger and M. Weil, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, 65–67 CrossRef PubMed.
- K. Nozaki, K. Takahashi, K. Nakano, T. Hiyama, H.-Z. Tang, M. Fujiki, S. Yamaguchi and K. Tamao, Angew. Chem., Int. Ed., 2003, 42, 2051–2053 CrossRef PubMed.
- H. Xu, K. Yin and W. Huang, Chem. – Eur. J., 2007, 13, 10281–10293 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem, Inc., Shawnee Mission, KS, 2009 Search PubMed.
- CrysAlisPro, Rigaku Oxford Diffraction, Kemsing, Sevenoaks, England, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra; S0 → S1 NTO; DSC, TGA, and CV data; PL and EL spectra. CCDC 1568697. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8tc03537g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |