
Electronic properties of isoindigo-based conjugated polymers bearing urea-containing and linear alkyl side chains†
 b
and
Simon
Rondeau-Gagné
*a
b
and
Simon
Rondeau-Gagné
*a
Abstract
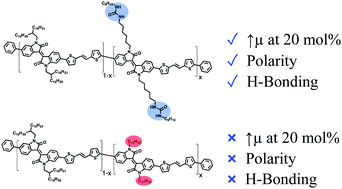
A side-chain engineering study has been performed with isoindigo-based conjugated polymers to modulate their physical and electronic properties through the incorporation of urea functionalities. Easily accessible and versatile, the incorporation of urea moieties in the side chains of conjugated polymers enables the formation of intermolecular hydrogen bonds between the polymer chains, resulting in a significant effect on the solid-state morphology and electronic properties. Incorporation of 0 to 20 mol% of urea-containing side chains was achieved, and the resulting materials were characterized by several techniques. Moreover, a comparison was also performed with similar polymers incorporating 0 to 20 mol% of dodecyl linear side chains. The organic field-effect transistors fabricated from the new materials showed that, at low content of urea moieties, the formation of hydrogen bonds did not allow for a significant improvement of the charge carrier mobility. However, devices made from the polymer with 20 mol% urea moieties showed an enhanced average charge mobility (0.032 cm2 V−1 s−1), in contrast to the polymer with 20 mol% dodecyl side chains (0.0073 cm2 V−1 s−1). The results obtained from the investigation of urea-containing isoindigo-based polymers confirm that the incorporation of urea moieties in polyisoindigos is a promising strategy to control the nanoscale morphology, solubility and polarity of conjugated polymers without reducing their performance in organic field-effect transistors.
- This article is part of the themed collection: Journal of Materials Chemistry C Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

