
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intermolecular interactions in molecular crystals and their effect on thermally activated delayed fluorescence of helicene-based emitters†
Anastasia
Klimash
 a,
Piotr
Pander
b,
Wim T.
Klooster
c,
Simon J.
Coles
c,
Przemyslaw
Data
b,
Fernando B.
Dias
*b and
Peter J.
Skabara
*a
a,
Piotr
Pander
b,
Wim T.
Klooster
c,
Simon J.
Coles
c,
Przemyslaw
Data
b,
Fernando B.
Dias
*b and
Peter J.
Skabara
*a
aWestCHEM, School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: peter.skabara@glasgow.ac.uk
bDepartment of Physics, Durham University, Durham, DH1 3LE, UK
cSchool of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK
First published on 12th September 2018
Abstract
Here, we discuss the influence of the crystal structure on the photophysical properties of two new TADF emitters containing a non-planar helical moiety. The presence of solvent in the crystal lattice of a diaza[5]helicene-based compound alters molecular packing significantly and suppresses aggregation. This results in more intense TADF emission and an increase in PLQY. Solution-processed OLED devices gave a maximum external quantum efficiency of 7.1%.
Introduction
The fabrication of OLED devices is one of the most mature and commercially successful applications of organic electronics. Yet, increasing the efficiency of OLED emitters remains the central subject of the research in the field. Organic fluorescent emitters can utilise only singlet excited states, since triplets are often non-emissive at room temperature. However, upon electrical excitation triplets and singlets are formed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio,1 thus the internal quantum efficiency (IQE) of fluorescent devices is limited to 25%. The use of thermally activated delayed fluorescence (TADF) materials is one of the most popular ways of promoting triplet harvesting, achieving internal quantum efficiency of up to 100% in devices based on purely organic compounds.2 In TADF materials the lowest triplet excited states (T1) are converted into the lowest singlet excited states (S1) via a reverse intersystem crossing (rISC) mechanism. The efficiency of this mechanism increases when the energy gap between S1 and T1 (ΔEST) is sufficiently small (i.e. less than 0.1 eV). However, the presence of a small ST gap is necessary but not sufficient for efficient TADF to be observed. In most TADF materials the ΔEST gap is reduced by minimising the overlap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) using donor–acceptor (D–A) molecules where the HOMO and LUMO are located on the electron donor and electron acceptor respectively, and the separation of orbitals is enhanced by introducing a π-bridge,3 or creating orthogonal donor and acceptor moieties, which have great influence on the TADF mechanism.2
:3 ratio,1 thus the internal quantum efficiency (IQE) of fluorescent devices is limited to 25%. The use of thermally activated delayed fluorescence (TADF) materials is one of the most popular ways of promoting triplet harvesting, achieving internal quantum efficiency of up to 100% in devices based on purely organic compounds.2 In TADF materials the lowest triplet excited states (T1) are converted into the lowest singlet excited states (S1) via a reverse intersystem crossing (rISC) mechanism. The efficiency of this mechanism increases when the energy gap between S1 and T1 (ΔEST) is sufficiently small (i.e. less than 0.1 eV). However, the presence of a small ST gap is necessary but not sufficient for efficient TADF to be observed. In most TADF materials the ΔEST gap is reduced by minimising the overlap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) using donor–acceptor (D–A) molecules where the HOMO and LUMO are located on the electron donor and electron acceptor respectively, and the separation of orbitals is enhanced by introducing a π-bridge,3 or creating orthogonal donor and acceptor moieties, which have great influence on the TADF mechanism.2
Despite the increasing amount of TADF materials reported to date, very few studies are focused on investigating TADF behaviour in pure solid state, i.e. without dispersing the emitter in a host. Recently, several TADF emitters with mechanochromism were reported,4 and the detailed analysis of the emission in different states for such compounds was presented.5 Some other studies were focused on understanding the role of the solid state solvation in TADF emitters.6,7 It is also crucial to understand the TADF processes in the solid state for compounds exhibiting aggregation-induced emission.8 Apart from that, the study of prompt and delayed fluorescence in crystals would be beneficial for extending the application of TADF phenomena to organic light-emitting transistors9 as crystalline materials have the potential to provide a rare combination of high charge carrier mobility and high luminescence.10
In this work we discuss the photophysical properties of two new helicene-based compounds in the crystalline phase. Mono- and diaza[5]helicenes are the acceptors of choice that provide necessary steric hindrance with their distorted structures. Phenoxazine is used as a donor. Its morpholine-like central ring fused to two benzene rings contributes to a higher twist within the structure, better separation of HOMO and LUMO, and induced CT transition.11
Helicenes belong to a family of polycyclic aromatic hydrocarbons (PAHs). They consist of several ortho-fused aromatic rings, and their non-planar chiral structure is the result of intramolecular steric repulsion rather than the presence of an asymmetric centre. Due to a twist, the benzene rings in helicenes are not planar hexagons, as the lengths of the bonds in the inner helix are bigger than those in the outer helix.12 However, π-electrons can still be delocalised through the distorted structure.13 Helicenes have high specific optical rotation,14 their skeleton is less rigid, and they are more soluble compared to planar PAHs.15 Such particularity of the structure encouraged the application of helicenes in a variety of fields ranging from asymmetric catalysis16,17 to chemical sensors,18–20 nonlinear optics,21,22 and circularly polarised luminescent materials.23,24 Photoluminescence quantum yields (PLQY) of unsubstituted helicenes are quite low,25 due to a fast intersystem crossing rate, which is probably the reason why they were not considered as suitable emissive materials for OLEDs until recently.26 However, the emissive properties of helicenes can be tuned by introducing various substituents.27 Their distorted structure that leads to a reduced π-conjugation and relatively large HOMO–LUMO gap even in higher helicenes makes them suitable candidates for blue OLEDs.28–30 Some theoretical studies also reveal the influence of spin–orbit coupling enhanced by the nonplanarity of the structure on the photophysical properties of helicenes.31,32 Others demonstrate that the chirality of the compounds has a significant effect on the behaviour of the bulk material, and the use of enantiopure and racemic mixtures can give drastically different results.33 All this proves that helicenes are a promising and exciting class of material for use in organic electronics, and a more in-depth understanding of the photophysical behaviour of these compounds is needed.
Results and discussion
Synthesis and characterisation
The key materials in this work are H1-PXZ and H2-PXZ and their synthesis is summarised in Scheme 1. For the preparation of H1-PXZ and H2-PXZ the corresponding bromides 1 and 2 were converted into phosphonium salts 3 and 4, which were next used for the synthesis of alkenes 6 and 7via Wittig olefination with aldehyde 5. Cores 8 and 9 were prepared by oxidative photocyclisation under UV irradiation.34 The preparation of helical acceptor cores is the limiting step for the scale-up of the materials as highly diluted solutions were used in order to avoid side products of [2+2] cycloaddition. The final compounds were prepared via Buchwald–Hartwig coupling between cores 8 and 9 with phenoxazine. The final step was carried out in a microwave synthesiser. Both H1-PXZ and H2-PXZ have good solubility in common solvents such as methanol, chloroform, dichloromethane, ethyl acetate and toluene. Full experimental details are given in the ESI† section.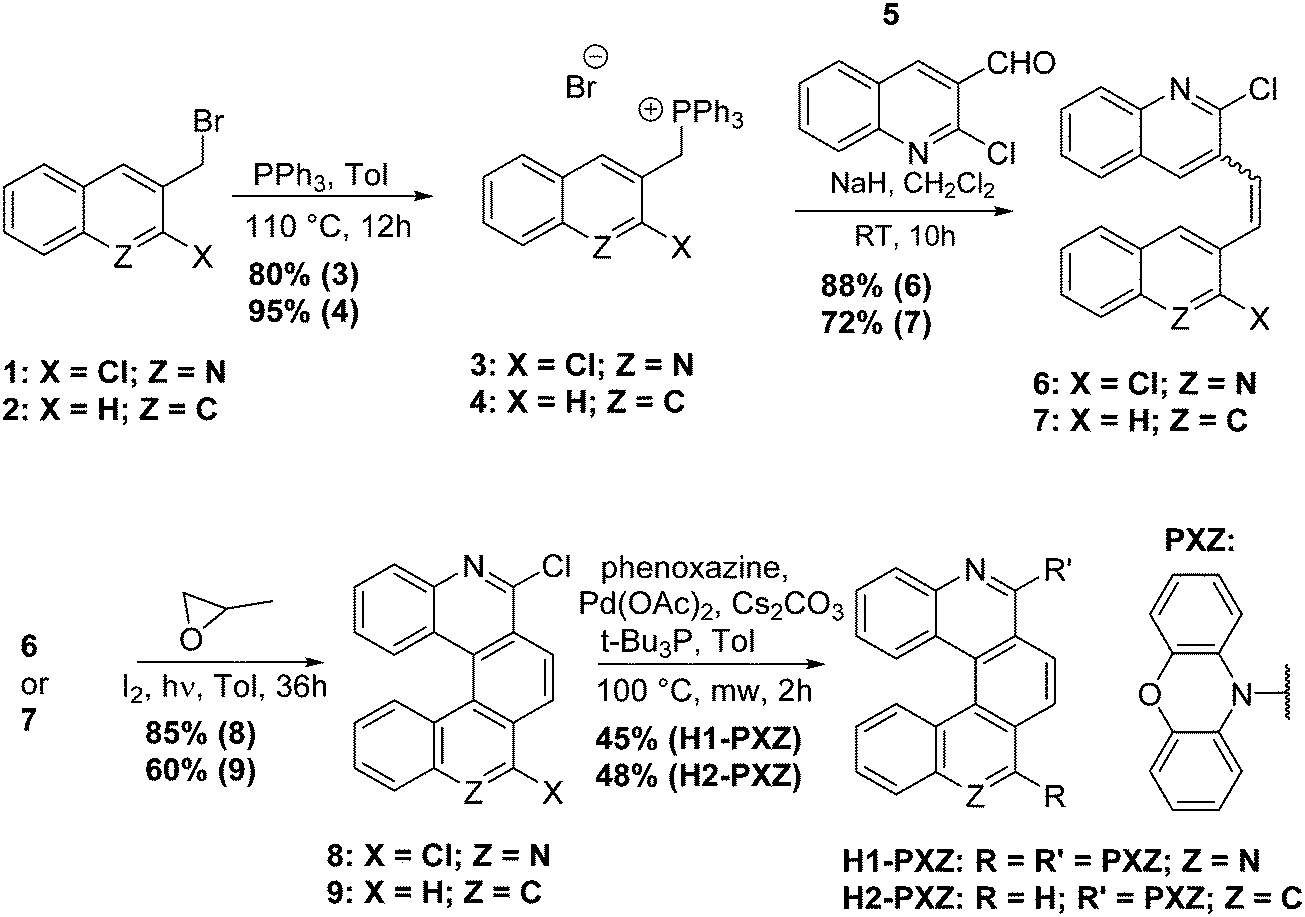 | ||
| Scheme 1 The synthesis of H1-PXZ and H2-PXZ. | ||
The molecular structure of H1-PXZ and H2-PXZ was confirmed by 1H NMR, 13C NMR and high-resolution mass spectrometry (Fig. S2 and S3, ESI†). The 1H NMR spectra of both compounds at room temperature display broad peaks corresponding to protons of phenoxazine moieties. Variable temperature NMR experiments (Fig. 1) demonstrate separation and further sharpening of peaks at lower temperature, which most likely indicates the presence of two conformers existing due to a restricted rotation between donor and acceptor. This is also consistent with the results of temperature-dependent photophysical studies described below. As expected, at higher temperatures the peaks coalesce.
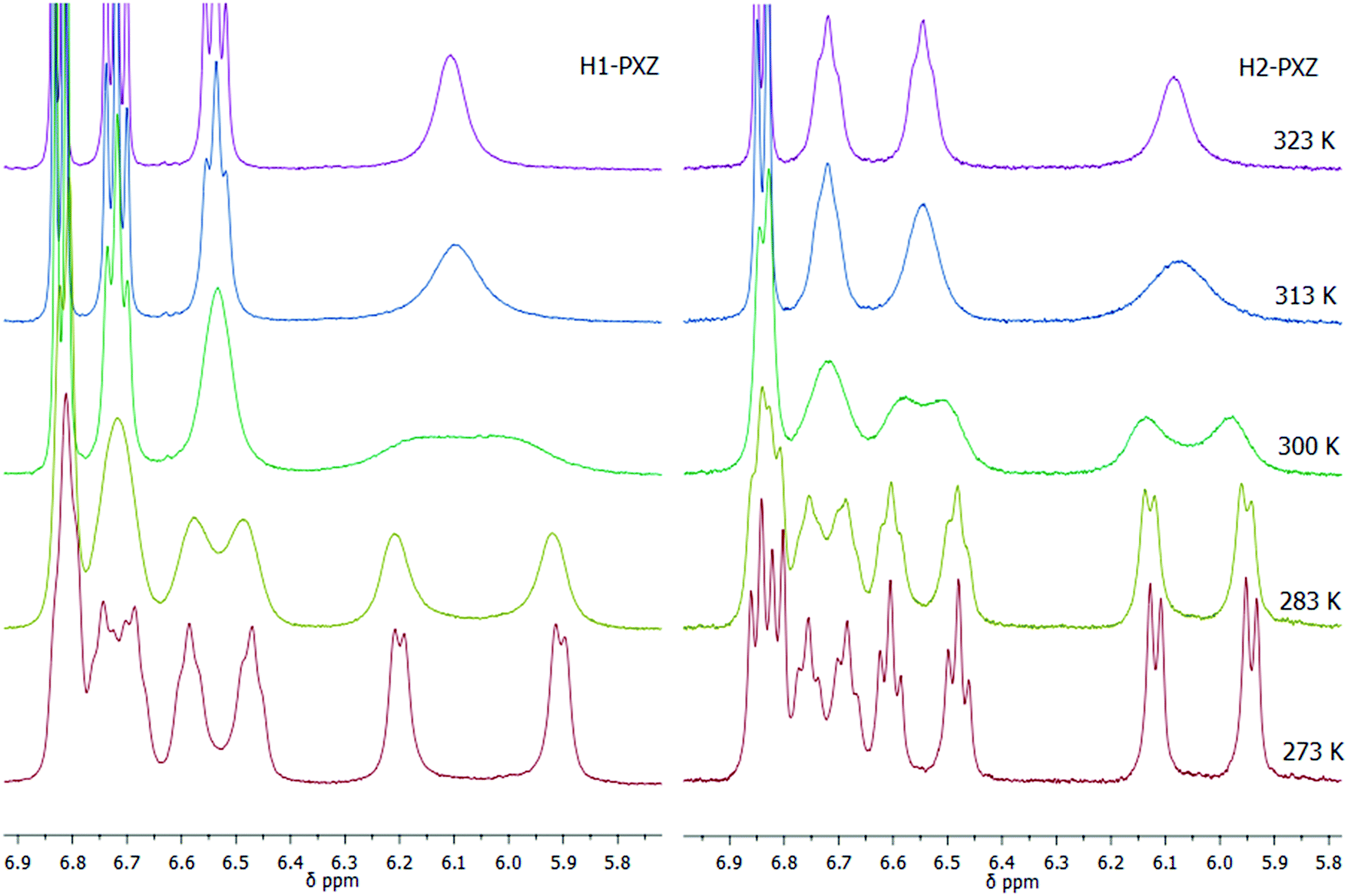 | ||
| Fig. 1 Variable temperature 1H NMR experiments for H1- and H2-PXZ at 273–323 K in chloroform-d, showing the aromatic peaks in the range 5.8 to 6.9 ppm. | ||
X-ray crystallography
The structures of both compounds were further confirmed by single crystal X-ray diffraction studies. Crystals of both H1-PXZ and H2-PXZ were obtained by slow evaporation from the mixture of ethyl acetate:hexane (1:7). For H1-PXZ two types of crystals were formed simultaneously: luminescent orange blocks (H1-PXZ-o) and non-luminescent red needles (H1-PXZ-r). In each case, compounds are present as a racemic mixture of two enantiomers. The torsion angles between the donor and acceptor moieties in H1-PXZ-o are reduced compared to H1-PXZ-r (Fig. 2 and Fig. S6.1, ESI†), being 73.1(2)°, and 88.0(1)°, respectively. The structure of H2-PXZ is shown in Fig. S6.1 (ESI†) and the molecule has a torsion angle of 82.9(3)°. Unlike the other two crystals, H1-PXZ-o contains hexane in the crystal lattice which significantly influences the packing within the crystal (Fig. S6.2a, ESI†) and also its photophysical behaviour. The molecular packing of H1-PXZ is the significant point of interest. In H1-PXZ-r (Fig. S6.2b, ESI†) we observe intermolecular π–π close contacts (ca. 3.2–3.5 Å) between the benzene rings of the phenoxazine units, whilst there are no close contacts for H1-PXZ-o due to the presence of the occluded hexane molecules. Since there are two phenoxazines per H1-PXZ molecule, this produces infinite chains of close contacts, which enhances the formation of low-emissive excimers.35 In H2-PXZ there is only one phenoxazine moiety, and this unit has a close intermolecular π–π interaction with an adjacent molecule to form aggregated pairs of molecules (Fig. S6.3, ESI†).
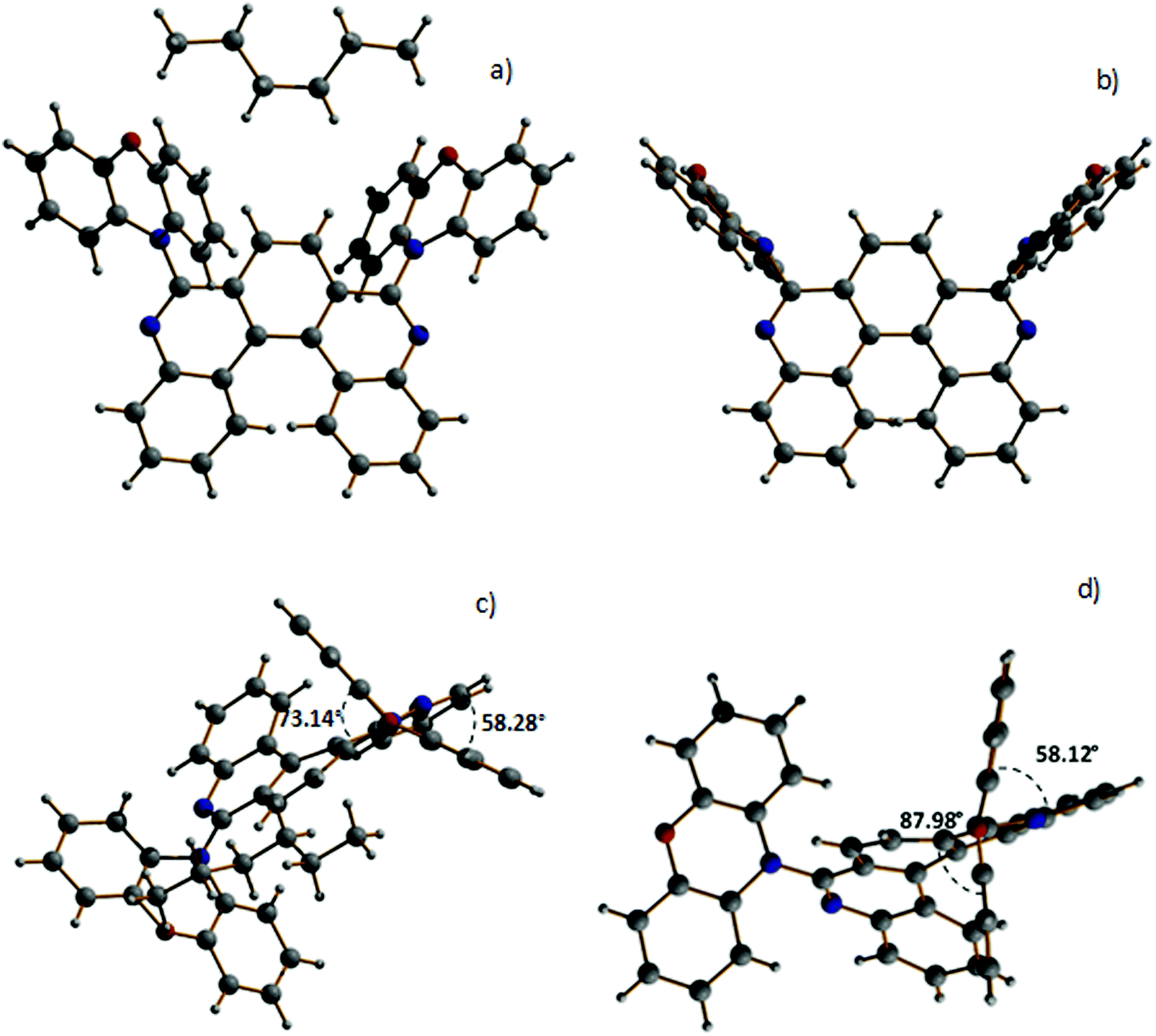 | ||
| Fig. 2 Molecular structures of H1-PXZ-o (a and c) and H1-PXZ-r (b and d). | ||
Electrochemical and thermal analysis
The electrochemistry data are summarised in Table S4.1 (ESI†). Both compounds demonstrate one reversible oxidation peak around 0.5 eV (Fig. S4.1, ESI†). H1-PXZ presents first reversible and second irreversible reduction peaks, while for H2-PXZ there is only an irreversible reduction peak (Fig. S4.1, ESI†).The thermal properties for both compounds were investigated in powder by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The DSC curves do not reveal any thermal events for H1- and H2-PXZ (Fig. S5.1, ESI†). Both compounds have high thermal stability and demonstrate 5% weight loss at 357 °C and 334 °C for H1-PXZ and H2-PXZ, respectively (Fig. S5.1, ESI†).
Solution state photophysical studies
The molecules presented in this work show the typical behaviour of D–A and D–A–D TADF molecules.36,37 This includes the existence of a charge transfer (CT) absorption band and the presence of local (LE) and CT photoluminescence in solution (Fig. 3). However, unlike other TADF compounds with a small S–T gap, a strong contribution of LE photoluminescence is observed in solution. This is probably related to a restricted rotation of donor moieties around the C–N axis, as it was observed in previous cases.38 The inability of the donor to quickly rotate around the C–N bond results in the donor–acceptor dihedral angle maintaining a nearly orthogonal orientation. This slows down the population of the CT state and allows the radiative decay rate from the LE state to compete with the formation of the CT state. However, the near orthogonal arrangement between D and A moieties cannot explain per se the absence of CT emission in solution. Other TADFs with similar arrangement show strong TADF in solution. Therefore, the cause of CT luminescence quenching is most probably due to internal conversion that more efficiently quenches the long-lived luminescence of the CT state, whereas the short-lived LE state is less affected. Molecule H1-PXZ, due to the strong electron-accepting properties of the acceptor, does show CT emission exclusively in non-polar solvents, such as methylcyclohexane (MCH) (Fig. 3). In other, more polar solvents the CT emission is quenched. However, this effect is also observed in many other TADFs.39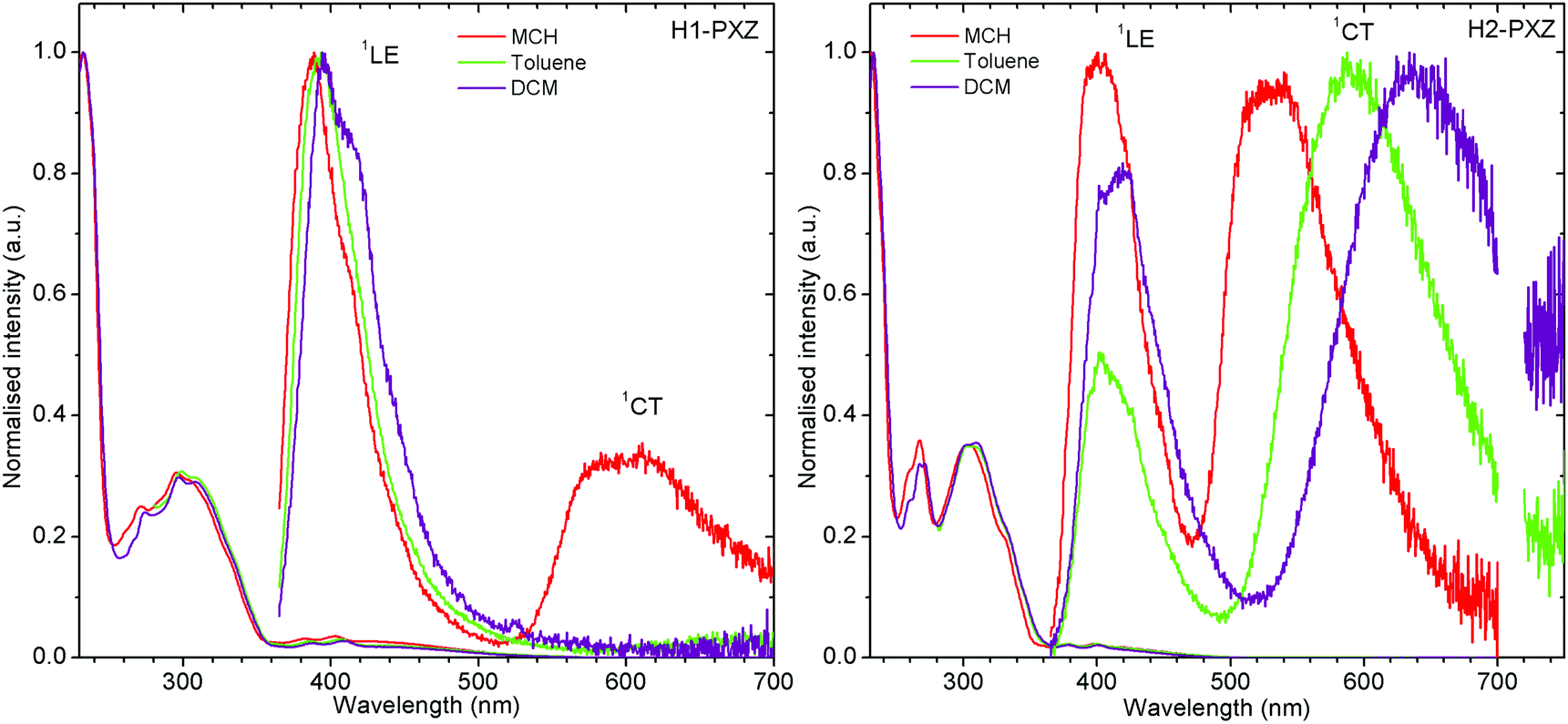 | ||
| Fig. 3 Photoluminescence spectra of H1-PXZ and H2-PXZ in solutions of various polarity. Note: CT emission of H1-PXZ is not observed in toluene and DCM, λex = 355 nm. | ||
Both molecules show a simultaneous LE and CT emission in non-polar solvent (Fig. 3), but the CT emission of H1-PXZ is completely quenched in toluene and DCM. On the other hand, the solvatochromic shift of CT emission in H2-PXZ can clearly be noted. The result suggests significantly stronger CT character of H1-PXZ relative to H2-PXZ. This results in the HOMO–LUMO orbitals of H1-PXZ being nearly completely orthogonal in the solvent because of orthogonality between donor and acceptor moieties.
Both molecules show delayed CT fluorescence in MCH solution (Fig. S7.1, ESI†) attributed to the TADF mechanism. Photoluminescence decay of H1-PXZ and H2-PXZ in solution at room temperature shows a typical TADF emission behaviour with two exponential regimes depicting prompt and delayed fluorescence, respectively. Note that the lifetime of both prompt and delayed fluorescence is shorter in solution than in the solid state (i.e. Fig. S7.1, Fig. 4b, 5 and Table S7.1, ESI†), suggesting a strong effect of non-radiative decay as proposed earlier in the text.
Photophysics in a polymer matrix
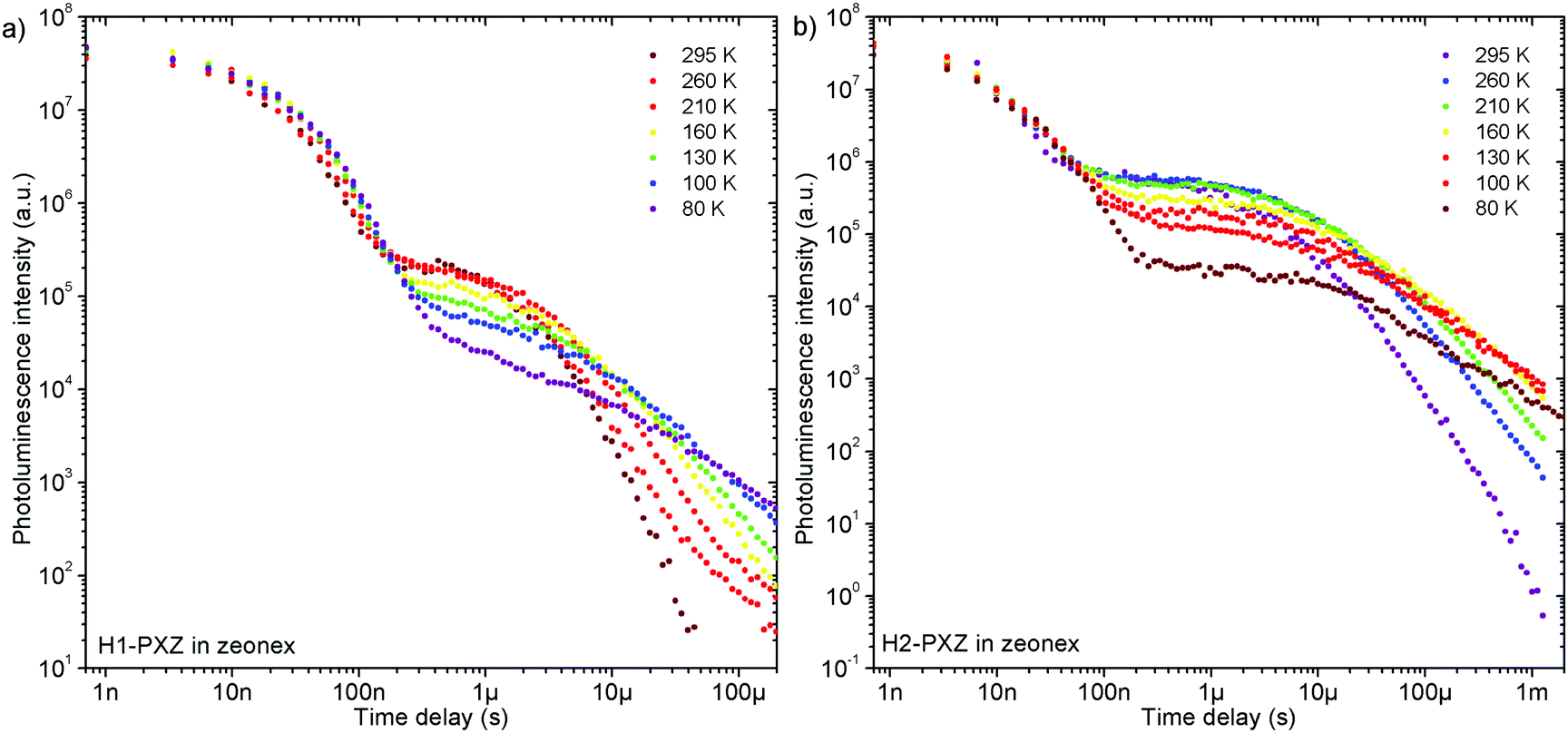
Remarkably, both molecules show TADF emission either doped in non-polar polymer Zeonex (Fig. 4a, b and 5) or MCH (Fig. S7.1, ESI†). In the solid state the non-radiative decay is suppressed, and CT emission is observed, and also TADF properties emerge. Note that in solution H2-PXZ seems to be a more efficient CT emitter than H1-PXZ (Fig. 3), which is probably due to faster radiative decay of the CT state that more effectively competes with internal conversion. For this reason, to fully understand these systems, we focus our investigations on the solid-state properties, i.e. in the amorphous film and in the crystal phase.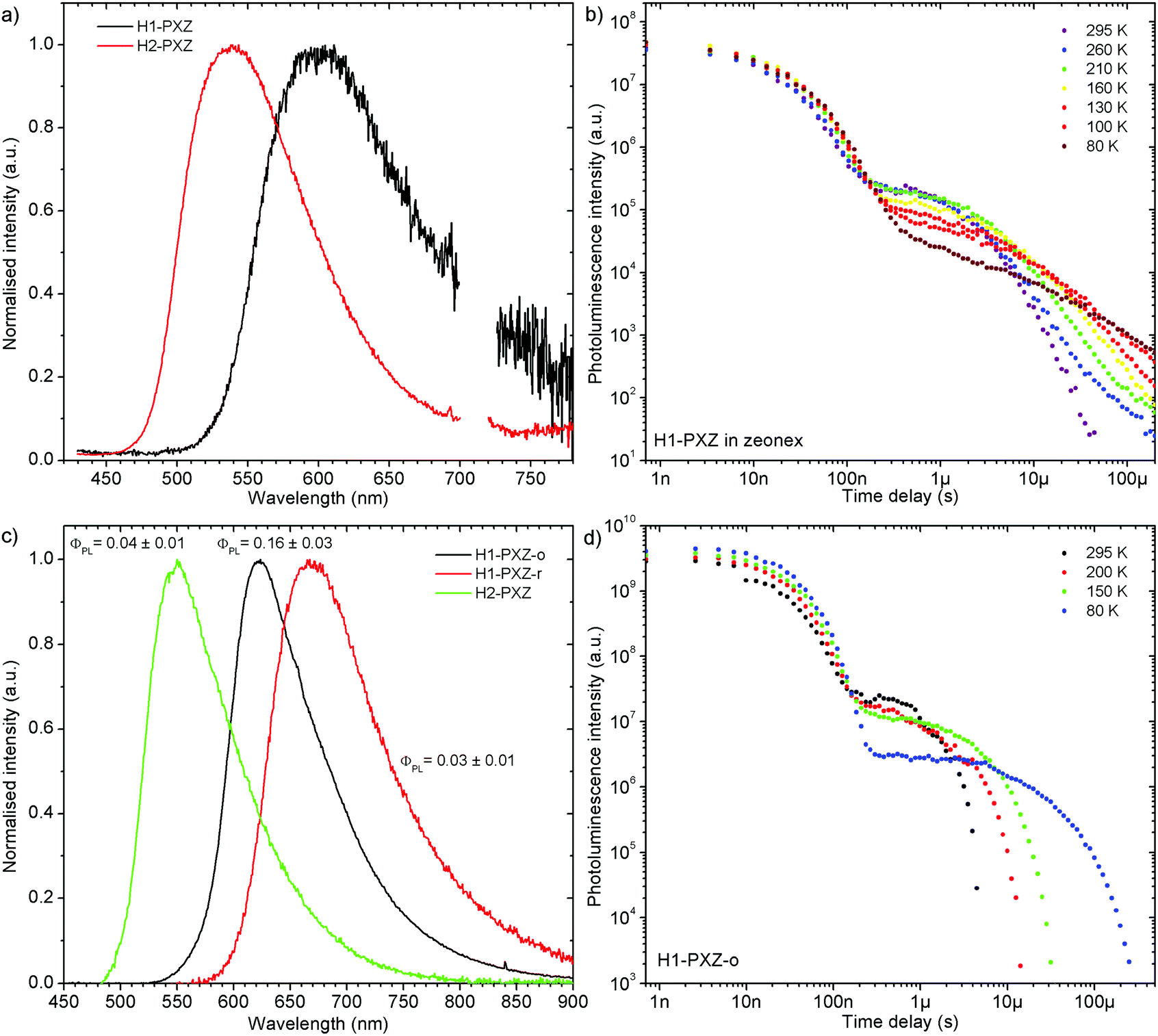 | ||
| Fig. 4 Photophysical characteristics of the pentahelicene derivatives in this study: (a) steady-state emission spectra in zeonex; (b) photoluminescence decay of H1-PXZ in zeonex; (c) steady-state photoluminescence spectra of molecular crystals; (d) photoluminescence decay of H1-PXZ-o crystals. | ||
 | ||
| Fig. 5 Photoluminescence decay of H1-PXZ and H2-PXZ in zeonex film (1% w/w) at various temperatures. | ||
The successful design of these molecules results in the singlet and triplet states being perfectly aligned even in non-polar surroundings, leading to a nearly zero S–T gap. In fact, in these systems phosphorescence is hardly observable even at 80 K, which makes the determination of the triplet state energy very difficult (Fig. S7.2 and S7.3, ESI†). Nevertheless, phosphorescence can relatively clearly be distinguished in H2-PXZ, indicating an S–T gap of 0.01 eV. In H1-PXZ the phosphorescence is not clearly observed, and the triplet energy cannot be determined directly from phosphorescence. The temperature dependence of TADF in these systems (Fig. S7.2c and S7.3c, ESI†) shows that the activation energy (Ea) is below 0.02 eV (12 ± 3 meV and 15 ± 3 meV in H1-PXZ and H2-PXZ, respectively); this indicates that the S–T gap in H1-PXZ is of the same order of magnitude as that of H2-PXZ. The increase in the delayed fluorescence intensity in the 300–200 K temperature range (Fig. S7.2c and S7.3c, ESI†), also shows that both H1-PXZ and H2-PXZ are influenced by non-radiative decay.
In H1-PXZ the prompt fluorescence (PF) and delayed fluorescence (DF) spectra are nearly identical and are only slightly blue-shifted at 80 K (Fig. S7.2a, ESI†). As was mentioned previously, due to a nearly zero S–T gap, observation of phosphorescence at 80 K is virtually impossible. Interestingly, at long delay (i.e. >100 ms) two emission components with comparative photoluminescence lifetime can be observed. One at 2.34 eV is identical to the CT emission of the molecule, thus should be attributed to the delayed fluorescence. The behaviour of the other component with ca. 0.5 eV higher energy resembles typical properties of phosphorescence in TADF molecules; however it is likely to originate from a different population of H1-PXZ molecules (Fig. S7.2b, ESI†). In such a case there is no communication between the two emissive states at 2.34 eV and at 2.84 eV, because they are located at different molecules. Such a situation is possible if in a rigidified polymer matrix at 80 K there is less freedom than at higher temperatures, and thus not all the molecules are able to adapt a conformation suitable for fast formation of a charge transfer state. The molecules which cannot adapt a suitable geometry thus emit phosphorescence from a local state (emission at 2.84 eV), whereas the others demonstrate only normal TADF CT emission (2.34 eV). Linear power dependence of the observed delayed fluorescence indicates its monomolecular origin (TADF) (Fig. S7.2d, ESI†).
The photophysical behaviour of H2-PXZ is more complicated than that of H1-PXZ as in the former the prompt fluorescence changes its spectrum over time (Fig. S7.3a, ESI†). The spectra recorded at 0.7 ns delay and 35 ns delay are different, with the latter being in fact more similar to the DF spectrum recorded at a longer delay time. This shows relaxation of the excited state molecular geometry. Delayed fluorescence originates from molecules with relaxed geometry, therefore the DF spectrum fits the so-called “late” PF spectrum. In the case of H2-PXZ it is possible to identify phosphorescence at long delays at 80 K (Fig. S7.3b, ESI†). This is because the phosphorescence spectrum is well resolved and clearly different from the CT emission. It is worth noting that TADF is still present at 80 K to some extent. Remarkably, the phosphorescence onset (2.57 eV) and relaxed CT emission onset (2.58 eV) are virtually isoenergetic. The lowest triplet state of the molecule (3LE) is likely to be located at the acceptor since the donor's phosphorescence is expected at higher energy. The power dependence of the delayed fluorescence is linear, which confirms the monomolecular origin of DF and in turn indicates TADF (Fig. S7.3d, ESI†).
Photophysics in crystals
Both zeonex films and solutions are isotropic and homogeneous environments, and due to the good solubility of both compounds in toluene, the formation of aggregates during film fabrication is unlikely. Moreover, the low concentration of both emitters used in these studies suppress bimolecular interactions between emitter molecules. On the contrary, in crystals of H1-PXZ and H2-PXZ the intermolecular interactions play a significant role by strongly suppressing the non-radiative decay, and by controlling the CT state energy. Typically, intermolecular interactions lead to aggregation induced quenching of luminescence.40 This results in H1-PXZ-r and H2-PXZ crystals exhibiting a low ΦPL. However, not only is the ΦPL low (0.03–0.04), but also the TADF emission arising from the crystals is weak at room temperature (Fig. 6 and 4c). In contrast, H1-PXZ-o crystals, that contain a molecule of hexane per each molecule of emitter, show an increased ΦPL = 0.16 ± 0.03 and clear presence of TADF (Fig. 4d). Comparing the crystals H1-PXZ-r without co-crystallised hexane molecules with H1-PXZ-o, the influence of the hexane molecule appears to be significant for the observation of TADF. Remarkably, the D–A dihedral angles in both crystals appear very close. For this reason, the differences observed in the TADF behaviour between the two H1-PXZ crystals should be attributed to the presence of hexane molecules in the crystal lattice itself. As discussed in the X-ray crystallography section, in H1-PXZ-r crystals the luminescence is suppressed due to the intermolecular π–π close contacts. The presence of the hexane molecules perturbs the molecular packing in the H1-PXZ-o crystals and minimises luminescence quenching occurring due to the aggregation. The H1-PXZ-o crystal also shows a higher CT state energy than H1-PXZ-r. Since there is no significant variation in the relative positions of the D–A units, this should be attributed to the change in dielectric constant of each crystal type. H1-PXZ as a D–A–D molecule shows a significant dipole moment even in the ground state, which gives rise to a larger dielectric constant in the solid state. In contrast, the presence of hexane may lead to a decrease in the dielectric constant, which results in the CT energy varying in each crystal. The variation of the CT energy not only changes the emission colour but also affects TADF. In H1-PXZ-o the TADF emission is clearly visible and observable even at low temperature (80 K), indicating an S–T gap close to zero. As the CT state energy becomes lower, the non-radiative decay increases, affecting both the lowest singlet (S1) and triplet (T1) states. This is known as the energy gap law. This concept explains the lower total ΦPL of H1-PXZ-r (a consequence of both S1 and T1 being affected by non-radiative decay), and much weaker, hardly observable TADF (indicating that the T1 state is affected).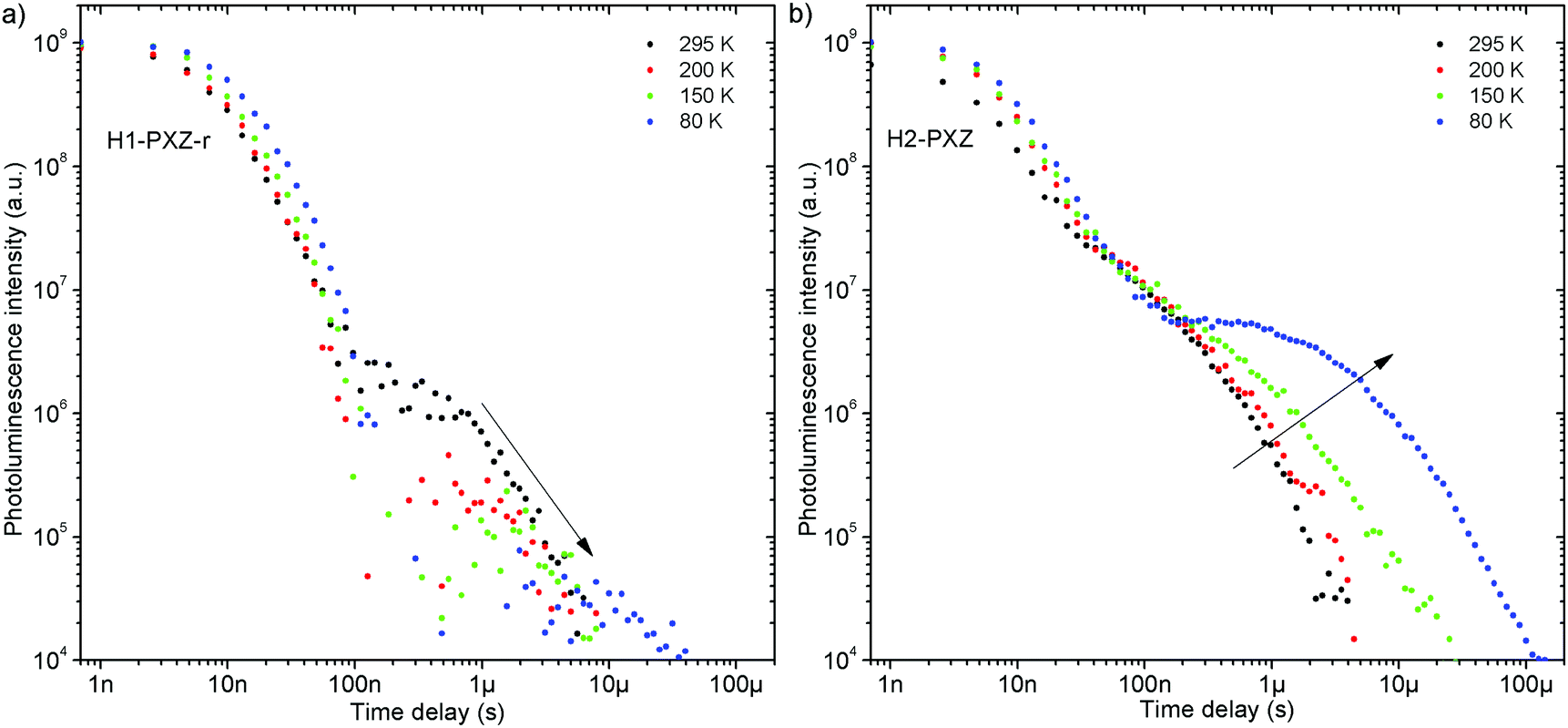 | ||
| Fig. 6 Photoluminescence decay of H1-PXZ-r and H2-PXZ crystals at various temperatures. | ||
H1-PXZ-r crystals, in comparison to H1-PXZ-o present a much lower photoluminescence quantum yield and so the delayed fluorescence is also very weak (Fig. 4d and 6a). However, the TADF clearly decreases in intensity and slows down with decrease in temperature in this case. On the other hand, H2-PXZ crystals show a negative temperature dependence of delayed fluorescence intensity, so the TADF intensity increases significantly at low temperature (Fig. 6b). This shows not only a significant influence of non-radiative decay, but also a very small S–T gap in the crystal.
H1-PXZ-o shows typical TADF properties at 295 K. The prompt and delayed fluorescence have the same emission spectrum and there is no evidence of the presence of any meta-stable conformers (Fig. 7a). At low temperature (Fig. 7b) one can note significant restriction of molecular motion, which results in the presence of at least two conformers, existing in short timescale (prompt fluorescence) and long timescale (delayed fluorescence). The signal observed in microsecond timescale is unlikely to be phosphorescence. This is evidenced by the lack of a third exponential component to be present in the photoluminescence decay (these would be: 1st prompt fluorescence, 2nd residual delayed fluorescence, and 3rd phosphorescence). As the TADF mechanism involves a competition between delayed fluorescence and phosphorescence, in the intermediate temperatures (between 295 and 80 K) there should be an additional exponential component present if phosphorescence was observed. This is however not observed as the decay consists of only two exponential regimes at all the temperatures investigated. Moreover, the phosphorescence is unlikely to be such a short-lived (i.e. 10–100 μs) emission without the presence of any heavy atoms.
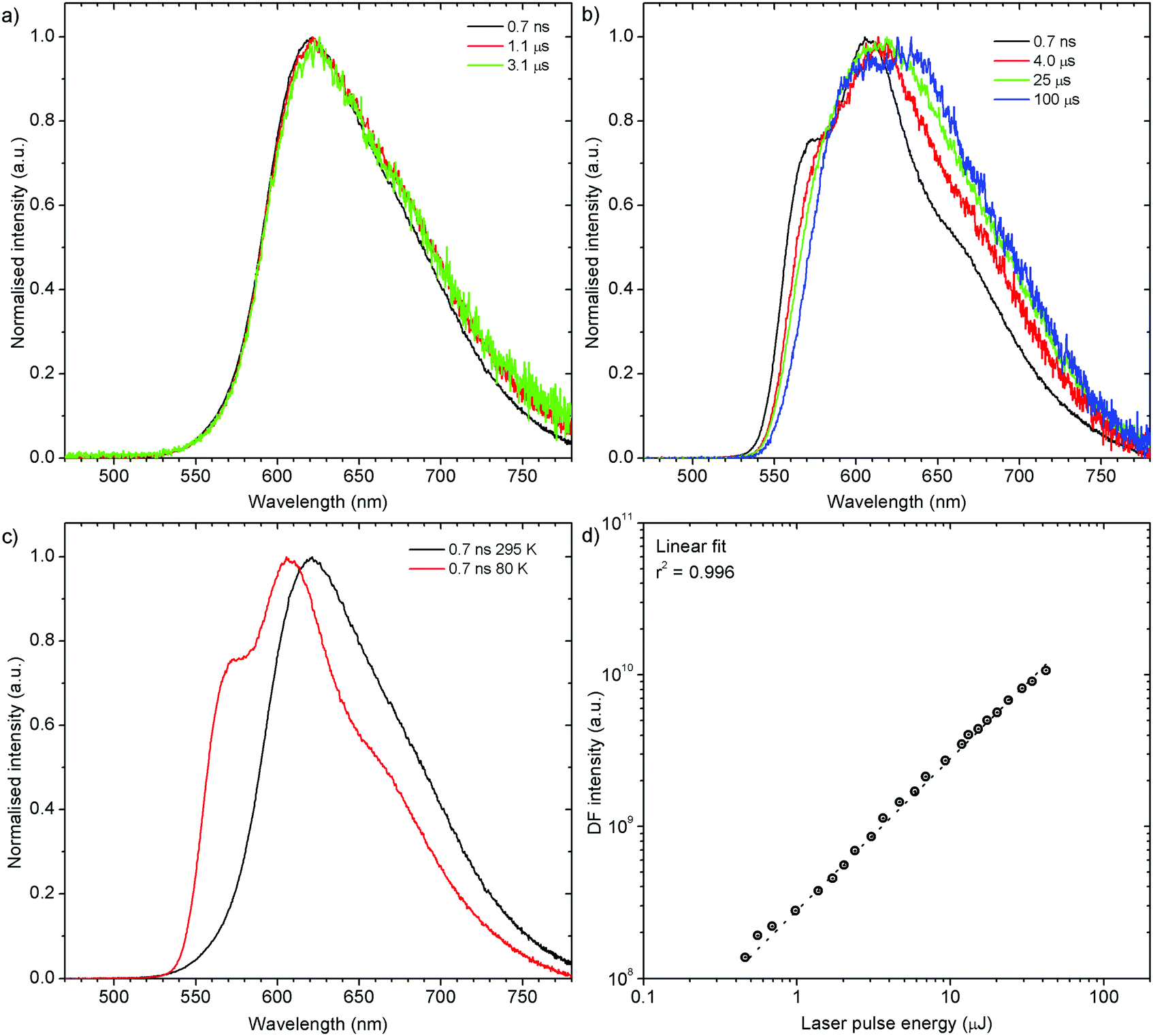 | ||
| Fig. 7 Photoluminescence properties of H1-PXZ-o crystals: (a) time-resolved spectra at 295 K; (b) time-resolved spectra at 80 K; (c) comparison of prompt fluorescence spectra at 295 K and 80 K; (d) power dependence of delayed fluorescence at 295 K. | ||
Prompt fluorescence spectra at 295 K and 80 K are clearly different (Fig. 7c), indicating a significant geometrical change in the singlet excited state at low temperatures. This shows clearly that the molecular motion is obstructed at low temperature, resulting in the singlet state not being able to rapidly relax its geometry. The power dependence experiment (Fig. 7d), proves that the delayed fluorescence recorded in H1-PXZ-o is a first order process, thus it is assigned as TADF.
H1-PXZ-r shows TADF emission properties at 295 K, but the delayed fluorescence is very weak. The prompt and delayed fluorescence have the same emission spectrum, and there are no conformers present (Fig. 8a). Similarly to H1-PXZ-o, in H1-PXZ-r at low temperature (Fig. 8b) one can note restriction of molecular motion. This results in the late prompt (i.e. 35 ns) and delayed fluorescence (microsecond region) to differ from early prompt fluorescence (i.e. 0.7 ns). We argue that this is due to restricted molecular motion and is a consequence of emission arising from two different populations of conformers. The signal observed in the microsecond timescale is unlikely to be phosphorescence, similarly to H1-PXZ-o and due to identical reasons.
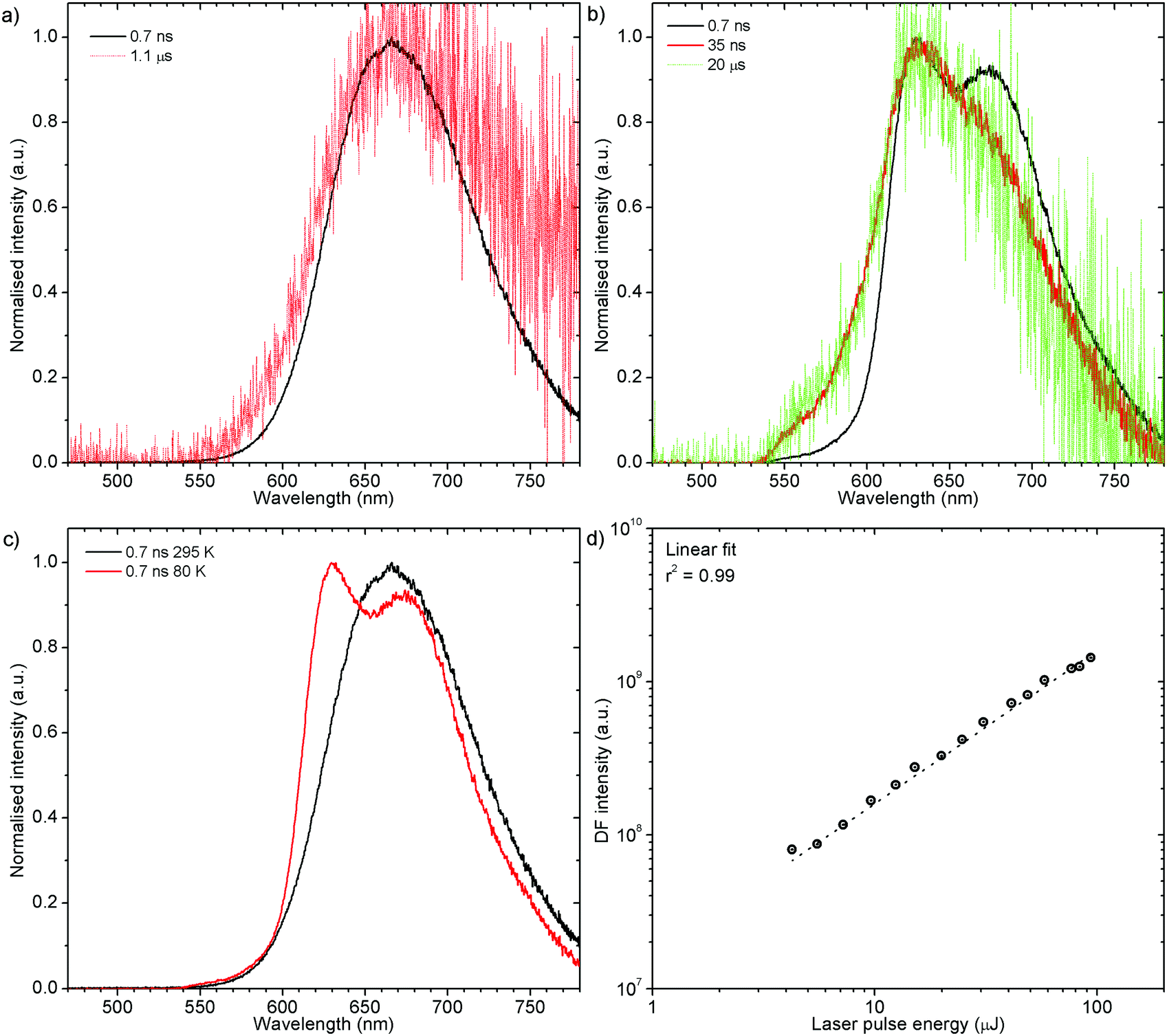 | ||
| Fig. 8 Photoluminescence properties of H1-PXZ-r crystals: (a) time-resolved spectra at 295 K; (b) time-resolved spectra at 80 K; (c) comparison of prompt fluorescence spectra at 295 K and 80 K; (d) power dependence of delayed fluorescence at 295 K. | ||
Prompt fluorescence spectra at 295 K and 80 K are clearly different (Fig. 8c) indicating a significant geometrical change in the singlet excited state at low temperatures. This shows clearly that the molecular motion is obstructed at low temperature, resulting in the singlet state not being able to rapidly relax its geometry. A power dependence experiment (Fig. 8d) proves the delayed fluorescence recorded in H1-PXZ-r is a first order process, thus it is TADF.
H2-PXZ shows TADF emission properties at 295 K, but the delayed fluorescence appears weak, yet seems to be stronger than in H1-PXZ-r crystals (Fig. 6b). The prompt and delayed fluorescence have the same emission spectrum and there are no conformers present (Fig. 9a). Similarly to H1-PXZ-o, and also in H2-PXZ at low temperatures (Fig. 9b), one can note restricted molecular motion. This results in the prompt fluorescence (0.7 ns) and delayed fluorescence (1 μs–1 ms) spectra to be different. We believe this is due to the same reasons as stated above in other types of molecular crystals investigated in this work. H2-PXZ crystals are, however, different from others discussed here. This is because at very late delay times, i.e. 79 ms, phosphorescence can clearly be observed (Fig. 9b). This is evidenced not only by the clearly vibronic shape of the emission spectrum but also by the fact that this emission appears to form a third exponential region (>50 ms), which is particularly different from the delayed fluorescence region (200 ns–1 ms) (Fig. S7.4, ESI†).
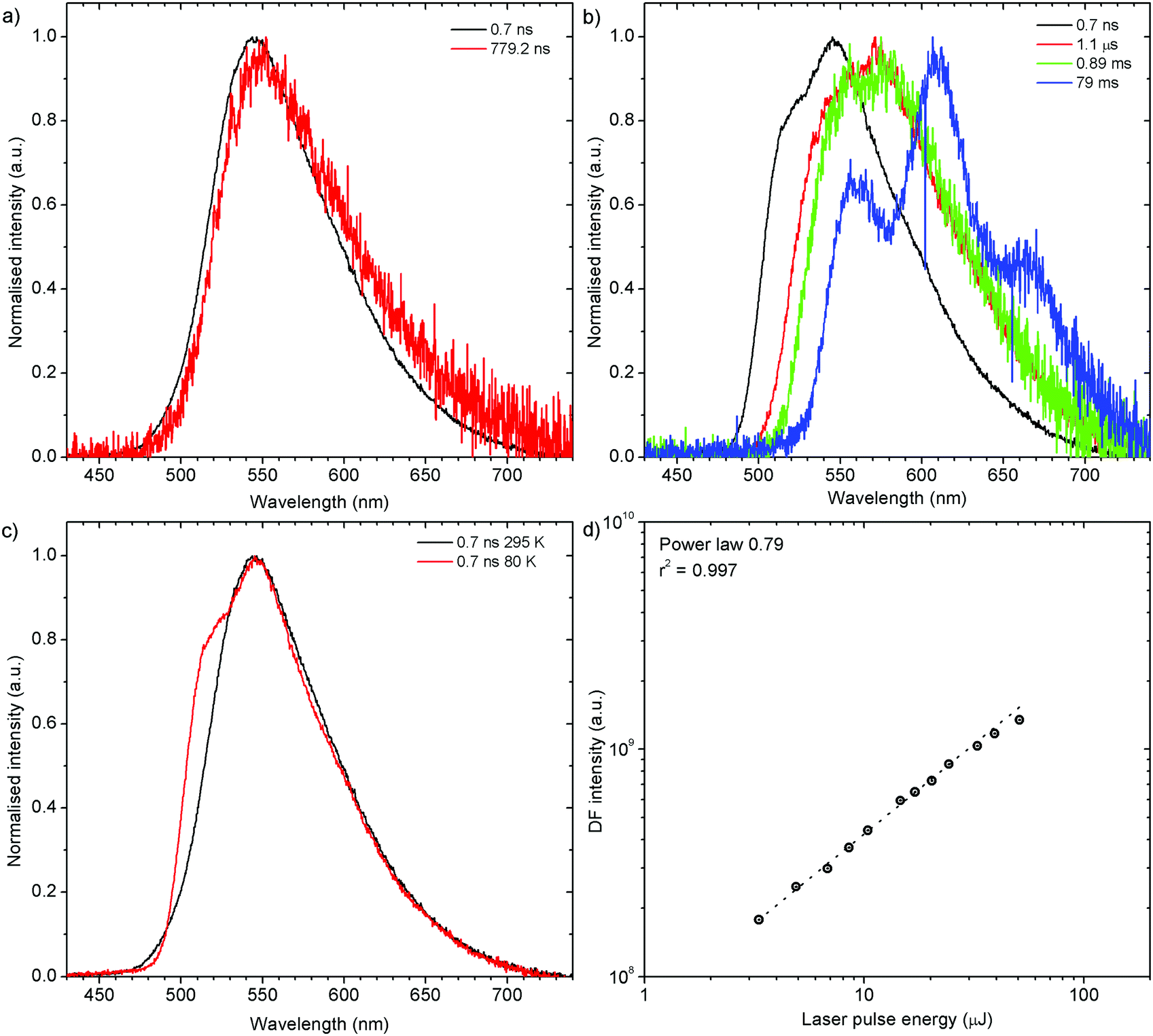 | ||
| Fig. 9 Photoluminescence properties of H2-PXZ crystals: (a) time-resolved spectra at 295 K; (b) time-resolved spectra at 80 K; (c) comparison of prompt fluorescence spectra at 295 K and 80 K; (d) power dependence of delayed fluorescence at 295 K. | ||
Prompt fluorescence spectra at 295 K and 80 K are clearly different (Fig. 9c), indicating a significant geometrical change in the singlet excited state at low temperatures, similar to the previous cases. Power dependence of delayed fluorescence in H2-PXZ crystals (Fig. 9d), is sublinear (power law 0.79). This likely indicates the monomolecular origin of delayed fluorescence but also an influence of an additional, exciton-quenching process. It is not clear what causes this effect, but one explanation can be the presence of singlet–singlet annihilation.
As the crystals differ in the photoluminescence quantum yield, the best parameter describing the amount of TADF produced by each type of crystal is the delayed to prompt fluorescence ratio (Fig. 10). In fact both H1-PXZ-o and H1-PXZ-r crystals show insignificant variation of the DF/PF with temperature. Over all temperatures the H1-PXZ-o shows visibly more TADF than H1-PXZ-r crystals. This could suggest that the H1-PXZ-o crystals show much better coupling of the CT state with the 3LE state, resulting in a larger triplet formation yield. On the contrary, H2-PXZ shows a large variation of the DF/PF ratio with temperature. At 80 K the DF/PF ratio is much larger than in the other cases. This means that the crystal is in fact a better TADF emitter than the other two discussed, but a strong non-radiative decay quenches triplet states much more effectively, so at room temperature the contribution of TADF is very little.
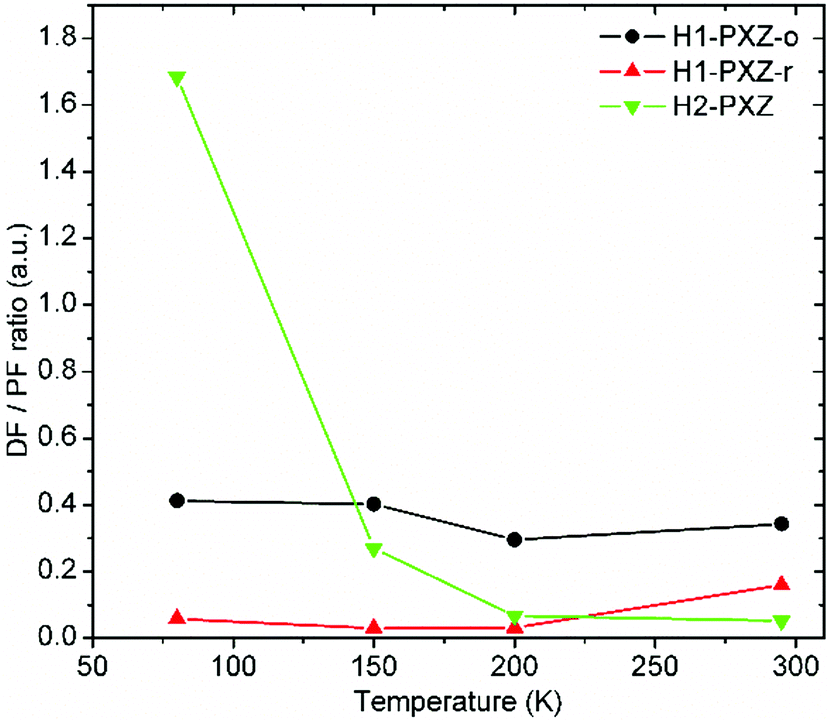 | ||
| Fig. 10 Delayed (DF) to prompt fluorescence (PF) ratio in the organic crystals as a function of temperature. See the ESI(†) section for further details of the interlayers. | ||
OLED devices
To evaluate the performance of the new materials for electroluminescence, solution-processed devices were fabricated using H1-PXZ (Dev 1) and H2-PXZ (Dev 2) as dopants in poly(N-vinylcarbazole):2-t-butylphenyl-5-biphenyl-1,3,4-oxadiazole (PVK:PBD, 60:40 w/w) as a mixed host. The orange (Dev 1), CIE (0.52, 0.41), and yellow (Dev 2), CIE (0.44, 0.50), emitting devices achieved 1.5% (Dev 1) and 7.1% (Dev 2) external quantum efficiency (EQE), and current efficiency (CE) of 2.7 cd A−1 and 13.9 cd A−1, respectively (Fig. 11 and Fig. S7.5, ESI†). The devices reach a maximum brightness of 1160 cd m−2 and 670 cd m−2 for Dev 1 and Dev 2, respectively. The turn-on voltage at 5 cd m−2 is lower for Dev 2, 5.1 V, than in Dev 1, 7.5 V. Interestingly, the efficiency roll-off of Dev 1 is clearly lower than in Dev 2. This might be associated with an unspecified degradation process in Dev 2.
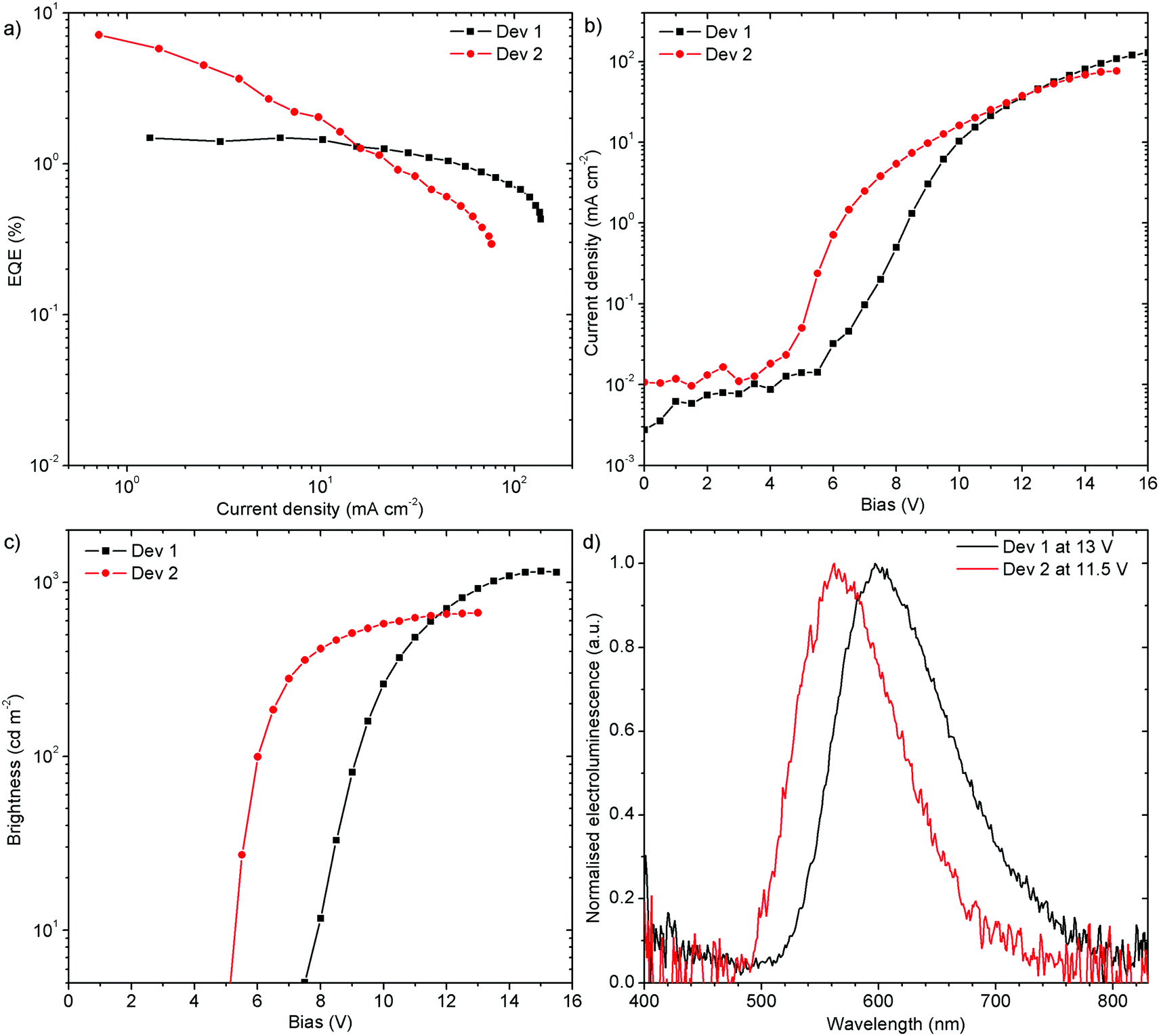 | ||
| Fig. 11 Electroluminescence characteristics of OLED devices fabricated using H1-PXZ and H2-PXZ as emitters: (a) EQE vs. current density; (b) current density vs. applied bias; (c) brightness vs. applied bias; (d) electroluminescence spectra. Device structure: ITO | HIL 1.3 N (45 nm) | PVKH (10 nm) | PVK:PBD (60:40) co 5% X (32 nm) | TPBi (50 nm) | LiF (0.8 nm) | Al (100 nm). Where X = H1-PXZ (Dev 1), X = H2-PXZ (Dev 2). | ||
Conclusions
To conclude, we have prepared two new helicene-based D–A–D and D–A compounds that demonstrate TADF in both the solid state and in solution. The photophysical study of crystals reveals that for H1-PXZ the presence of hexane in the crystal lattice increases not only the PLQY, but also the intensity of TADF emission. Thus, both parameters are influenced much more by the packing of the molecules in the lattice than by the relative position of donor and acceptor moieties within the molecule.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement H2020-MSCA-ITN-2015/674990 project “EXCILIGHT”. The data for the work can be accessed from http://dx.doi.org/10.5525/gla.researchdata.634 and CCDC 1853153, 1853154 and 1853155 contains supplementary X-ray crystallographic data for H1-PXZ-o, H1-PXZ-r and H2-PXZ, respectively.† We acknowledge the EPSRC UK National Mass Spectrometry Facility (NMSF) at Swansea University for HRMS.41References
- A. Köhler and H. Bässler, Mater. Sci. Eng., R, 2009, 66, 71–109 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- S. Y. Lee, T. Yasuda, I. S. Park and C. Adachi, Dalton Trans., 2015, 44, 8356–8359 RSC.
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC.
- R. Pashazadeh, P. Pander, A. Lazauskas, F. B. Dias and J. V. Grazulevicius, J. Phys. Lett., 2018, 1172–1177 CAS.
- B. L. Cotts, D. G. McCarthy, R. Noriega, S. B. Penwell, M. Delor, D. D. Devore, S. Mukhopadhyay, T. S. De Vries and N. S. Ginsberg, ACS Energy Lett., 2017, 2, 1526–1533 CrossRef CAS.
- T. Northey, J. Stacey and T. J. Penfold, J. Mater. Chem. C, 2017, 5, 11001–11009 RSC.
- J. Guo, X.-L. Li, H. Nie, W. Luo, S. Gan, S. Hu, R. Hu, A. Qin, Z. Zhao, S.-J. Su and B. Z. Tang, Adv. Funct. Mater., 2017, 27, 1606458 CrossRef.
- L. Song, Y. Hu, Z. Liu, Y. Lv, X. Guo and X. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 2711–2719 CrossRef CAS PubMed.
- J. Liu, H. Zhang, H. Dong, L. Meng, L. Jiang, L. Jiang, Y. Wang, J. Yu, Y. Sun, W. Hu and A. J. Heeger, Nat. Commun., 2015, 6, 10032 CrossRef CAS PubMed.
- P. Borowicz, J. Herbich, A. Kapturkiewicz, M. Opallo and J. Nowacki, Chem. Phys., 1999, 249, 49–62 CrossRef CAS.
- R. Kuroda, J. Chem. Soc., Perkin Trans. 2, 1982, 789–794 RSC.
- J. M. Schulman and R. L. Disch, J. Phys. Chem. A, 1999, 103, 6669–6672 CrossRef CAS.
- R. H. Martin and M. J. Marchant, Tetrahedron, 1974, 30, 343–345 CrossRef CAS.
- T. J. Katz, L. Liu, N. D. Willmore, J. M. Fox, A. L. Rheingold, S. Shi, C. Nuckolls and B. H. Rickman, J. Am. Chem. Soc., 1997, 119, 10054–10063 CrossRef CAS.
- T. Kawasaki, K. Suzuki, E. Licandro, A. Bossi, S. Maiorana and K. Soai, Tetrahedron: Asymmetry, 2006, 17, 2050–2053 CrossRef CAS.
- K. Yavari, P. Aillard, Y. Zhang, F. Nuter, P. Retailleau, A. Voituriez and A. Marinetti, Angew. Chem., Int. Ed., 2014, 53, 861–865 CrossRef CAS PubMed.
- D. J. Weix, S. D. Dreher and T. J. Katz, J. Am. Chem. Soc., 2000, 122, 10027–10032 CrossRef CAS.
- D. Z. Wang and T. J. Katz, J. Org. Chem., 2005, 70, 8497–8502 CrossRef CAS PubMed.
- M. T. Reetz and S. Sostmann, Tetrahedron, 2001, 57, 2515–2520 CrossRef CAS.
- T. Verbiest, S. V. Elshocht, M. Kauranen, L. Hellemans, J. Snauwaert, C. Nuckolls, T. J. Katz and A. Persoons, Science, 1998, 282, 913–915 CrossRef CAS PubMed.
- B. J. Coe, D. Rusanova, V. D. Joshi, S. Sánchez, J. Vávra, D. Khobragade, L. Severa, I. Císařová, D. Šaman, R. Pohl, K. Clays, G. Depotter, B. S. Brunschwig and F. Teplý, J. Org. Chem., 2016, 81, 1912–1920 CrossRef CAS PubMed.
- J. E. Field, G. Muller, J. P. Riehl and D. Venkataraman, J. Am. Chem. Soc., 2003, 125, 11808–11809 CrossRef CAS PubMed.
- K. Nakamura, S. Furumi, M. Takeuchi, T. Shibuya and K. Tanaka, J. Am. Chem. Soc., 2014, 136, 5555–5558 CrossRef CAS PubMed.
- J. B. Birks, D. J. S. Birch, E. Cordemans and E. V. Donckt, Chem. Phys. Lett., 1976, 43, 33–36 CrossRef CAS.
- S. Sahasithiwat, T. Mophuang, L. Menbangpung, S. Kamtonwong and T. Sooksimuang, Synth. Met., 2010, 160, 1148–1152 CrossRef CAS.
- H. Kubo, T. Hirose and K. Matsuda, Org. Lett., 2017, 19, 1776–1779 CrossRef CAS PubMed.
- W. Hua, Z. Liu, L. Duan, G. Dong, Y. Qiu, B. Zhang, D. Cui, X. Tao, N. Cheng and Y. Liu, RSC Adv., 2015, 5, 75–84 RSC.
- T. Chen, B. Zhang, Z. Liu, L. Duan, G. Dong, Y. Feng, X. Luo and D. Cui, Tetrahedron Lett., 2017, 58, 531–535 CrossRef CAS.
- S. Sahasithiwat, T. Sooksimuang, L. Kangkaew and W. Panchan, Dyes Pigm., 2017, 136, 754–760 CrossRef CAS.
- K. Schmidt, S. Brovelli, V. Coropceanu, J.-L. Brédas, C. Bazzini, T. Caronna, R. Tubino and F. Meinardi, J. Phys. Chem. A, 2006, 110, 11018–11024 CrossRef CAS PubMed.
- K. Schmidt, S. Brovelli, V. Coropceanu, D. Beljonne, J. Cornil, C. Bazzini, T. Caronna, R. Tubino, F. Meinardi, Z. Shuai and J.-L. Brédas, J. Phys. Chem. A, 2007, 111, 10490–10499 CrossRef CAS PubMed.
- Y. Yang, B. Rice, X. Shi, J. R. Brandt, R. Correa da Costa, G. J. Hedley, D.-M. Smilgies, J. M. Frost, I. D. W. Samuel, A. Otero-de-la-Roza, E. R. Johnson, K. E. Jelfs, J. Nelson, A. J. Campbell and M. J. Fuchter, ACS Nano, 2017, 11, 8329–8338 CrossRef CAS PubMed.
- D. Waghray, J. Zhang, J. Jacobs, W. Nulens, N. Basarić, L. V. Meervelt and W. Dehaen, J. Org. Chem., 2012, 77, 10176–10183 CrossRef CAS PubMed.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CrossRef CAS PubMed.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612–2615 RSC.
- D. d. S. Pereira, P. L. dos Santos, J. S. Ward, P. Data, M. Okazaki, Y. Takeda, S. Minakata, M. R. Bryce and A. P. Monkman, Sci. Rep., 2017, 7, 6234 CrossRef PubMed.
- J. Mei, Y. Hong, W. Y. Lam Jacky, A. Qin, Y. Tang and Z. T. Ben, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1853153–1853155. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8tc03390k |
| This journal is © The Royal Society of Chemistry 2018 |