
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
n-Type organic electrochemical transistors: materials and challenges
Hengda
Sun
,
Jennifer
Gerasimov
,
Magnus
Berggren
and
Simone
Fabiano
 *
*
Laboratory of Organic Electronics, Department of Science and Technology, Linköping University, Norrköping SE-601 74, Sweden. E-mail: simone.fabiano@liu.se
First published on 31st August 2018
Abstract
Organic electrochemical transistors (OECTs) have emerged as an enabling technology for the development of a variety of applications ranging from digital logic circuits to biosensors and artificial synapses for neuromorphic computing. To date, most of the reported OECTs rely on the use of p-type (hole transporting) conducting and semiconducting polymers as the channel material, while electron transporting (n-type) OECTs are yet immature, thus precluding the realization of advanced complementary circuitry. In this highlight, we review and discuss recent achievements in the area of n-type OECTs, in particular targeting recently reported n-type channel materials and how these have enabled a considerable advancement of OECT circuit capabilities. Further, the critical challenges currently limiting the performance of n-channel OECTs are summarized and discussed, setting material design guidelines for the next generation n-type and complementary OECTs.
1. Introduction
Organic electrochemical transistors (OECTs) are in a stage of rapid development as novel applications that make use of these versatile devices continue to emerge. OECTs are characterized by the coupling of both ionic and electronic inputs to modulate transistor channel conductance. This attribute renders OECTs ideal for interfacing electronics with biological systems, which make use of ionic and biochemical currents and gradients for signalling.1,2 To date, OECTs have demonstrated their utility for molecular sensing,3–5 cell culture analysis,6–9 medical diagnostics,10,11 neuromorphic computing,12,13 digital logic circuits,14 and printed electronics15–17 on a variety of substrates including soft, low-cost, transparent, and conformable materials.18Until very recently, however, only p-type conducting and semiconducting polymers have been investigated as channel materials, which has severely hampered the development of the OECT technology. With n-type OECTs also at hand, twinned with the p-type ones, large-scale complementary circuits become possible, which promises for a more versatile OECT technology operating at a relatively lower static power consumption and with improved noise immunity. This bottle-neck has partly been in place for a long time, because of the many stringent and sometimes conflicting requirements to produce materials suitable for n-type conduction.19 Further restrictions apply when designing channel materials for OECTs, since the material must exhibit reversible charge accumulation and ion exchange within the safe potential range of the chosen electrolyte–solvent system. The material should also be chemically and physically stable in both the oxidized and reduced form, be stable during ion penetration into the channel volume and exhibit a sufficiently high electron mobility. If the above criteria are met, it is possible to produce all-OECT complementary circuits, which will carry all the benefits of a significant reduction in static power consumption required for device operation. Reducing power consumption would set the stage for the emergence of portable, battery-powered systems, large-surface sensor networks and RFID tags with extended operating range.20
In this highlight, we delve into the working principles of OECTs, provide an overview of recently reported n-type channel materials and how these have advanced the performance and capabilities of the OECT circuits, and identify the material design challenges that are to be addressed for the next developments in this field.
2. Working principle of OECTs
To date, most of the efforts applied to incorporating organic semiconductors into transistor structures have been focused on optimizing organic field-effect transistors (OFETs).21 A typical OFET comprises a solid dielectric layer sandwiched between a channel semiconductor material and a gate electrode (Fig. 1A). In an OFET, a gate-induced electrostatic charge buildup at the interface of the semiconductor and the gate dielectric leads to modulation of the channel conductance.22 If the solid gate dielectric is replaced by an electrolyte (Fig. 1B), ions move under the influence of the electric field towards the organic semiconductor (OSC) surface and lead to the formation of an electrical double layer (EDL), along the electrolyte–semiconductor interface.23 This modified OFET is referred to as an electrolyte-gated OFET (EGOFET) and has the advantage of exhibiting a significantly higher capacitance (typically of 1–10 μF cm−2), which allows for device operation at relatively lower gate voltages, usually below 2 V.24,25 If the channel material is permeable to ions, charge accumulation is no longer limited to the interface between the channel and the electrolyte only, and the device is then referred to as an OECT (Fig. 1C). Hence, the distinguishing feature of the OECTs is that ions can enter the channel material and change the electronic conductivity throughout its entire volume. In this configuration, individual polymer chains, or nano-crystallites thereof, provide a capacitive interface, thus leading to a volumetric capacitance which can be orders of magnitude larger than the EDL capacitance residing along the interface between an electrolyte and an organic semiconductor layer.26 Because of the high volumetric capacitance, traditional metal gate electrode may not provide enough charges to dope the OECT channel, necessitating the use of redox gate electrodes, most commonly Ag/AgCl, or electrodes with large enough specific surface area, such as porous carbon or carbon nanotubes.27 It is worth noting, however, that since OECT operation requires ion migration through a solid medium, the operating speeds of OECTs tend to be slow. Typical OECTs can operate only below the kHz frequency range28,29—much lower than the MHz range achieved by OFETs.30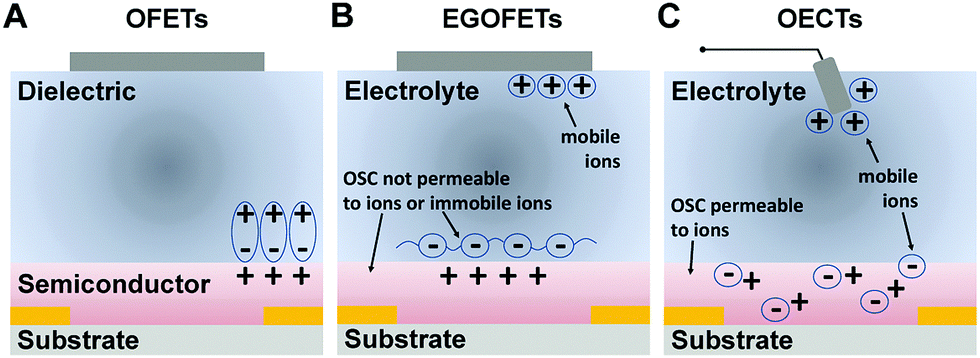 | ||
| Fig. 1 Device cross-section schematic showing the working principle of (A) OFETs, (B) EGOFETs, and (C) OECTs. | ||
The (semi)conducting nature of the channel material in the pristine state (i.e. in the absence of a gate voltage) dictates the operation mode of the OECT. When a highly conductive polymer, having an excess of free charge carriers, is used as the channel material, the OECT works in depletion mode. This is the case of OECTs including the well-known mixed ionic-electronic conductor poly(3,4-ethylenedioxythiophene) doped with poly(styrene sulfonate) (PEDOT:PSS) as the channel. At zero gate voltage, the transistor is in the on state as the sulfonate groups of the PSS compensate the mobile holes in the PEDOT. When a positive gate voltage is applied, holes in the channel are replaced by injected cations and the transistor switches off. In contrast, accumulation mode OECTs utilize semiconducting polymers that are undoped in their pristine state as the channel material. In the absence of gate voltage, the device is in the off state. When a gate voltage is applied, ions driven into the channel dope the film and increase its conductivity. In this case, mobile carriers are accumulated in the channel to turn the transistor on.
The most important figure of merit for an OECT device is the transconductance (gm), which is the device parameter relating the modulation of the drain current (ID) to the change in gate voltage (VG). The transconductance is then a direct measure of the extent to which an OECT can amplify an input signal and is extracted from the first derivative of transfer curve (gm = ∂ID/∂VG). The gm depends on both the channel geometry and biasing conditions, so one should make sure to compensate for device geometries (i.e., channel length L, channel width W, and film thickness d) when comparing the performance of OECT channel materials. A commonly used model, reported by Bernards et al.31 for depletion mode OECT, expresses gm in the saturation regime as
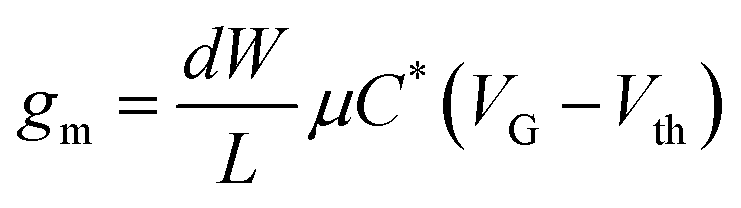
3. n-Type OECTs
3.1 n-Channel semiconductor materials: an OFET viewpoint
The development of high-performance organic semiconductors has traditionally been driven by the quest for efficient electron-transporting materials for OFETs. n-Channel polymers as well as molecular systems have historically lagged behind their p-type counterparts. As both theory and experiments have shown, the reason for this developmental delay is not due to intrinsic differences between the transport mechanisms for electrons and holes,34 but mainly due to the ambient instability of the electron-transporting materials. Typically, this instability in air arises from the susceptibility of the organic π-radical anions to react with atmospheric water and/or oxygen, an issue first discussed by de Leeuw et al.35 As the redox potential of H2O is −0.66 V vs. the saturated calomel electrode (SCE), it was reasoned that n-type organic semiconductors are stable at ambient atmospheric conditions only when their lowest unoccupied molecular orbital (LUMO) is deeper than −4.0 eV (Fig. 2).36 In addition to ambient stability, a low LUMO energy would also reduce the barrier height for the injection of electrons from commonly used high-work-function metal electrodes (e.g. Au). Thus, from an energetic perspective, the use of low-work-function metals, such as aluminium, should be preferable.37 However, oxidation of these conductor surfaces by air leads to the formation of an insulating layer, which eliminates the benefit of a lower energy barrier.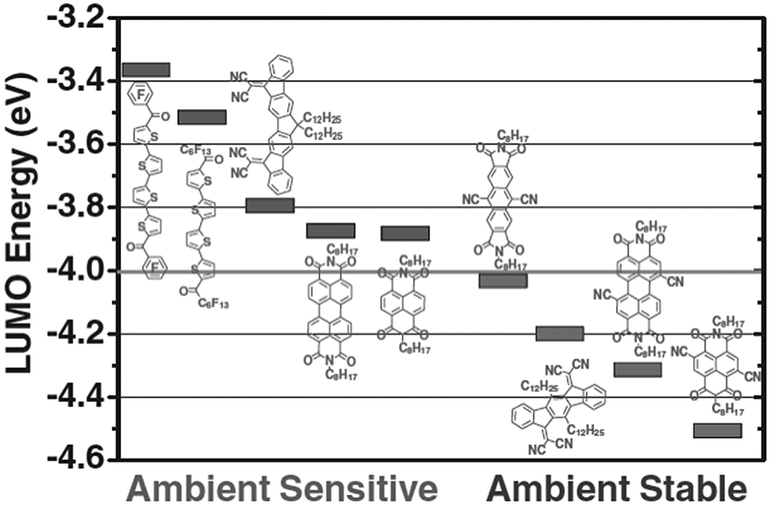 | ||
| Fig. 2 Correlation between LUMO energy and ambient stability for several organic n-type materials. Reprinted with permission from ref. 36 copyright 2011 Wiley-VCH. | ||
Numerous material design strategies have been developed to synthesize n-channel materials that satisfy this energetic requirement and a comprehensive description of the latest progress in n-type semiconductor synthesis—mostly for OFET applications—can be found in several recent reviews.38–42 For instance, advancements in air-stable n-channel materials for OFETs have been made possible by the development of electron-deficient building blocks, such as perylenediimides (PDI),43 naphthalenediimides (NDI),44 diketopyrrolopyrrole (DPP),45 isoindigo (IID),46 benzodifurandione (IBDF),47 to name just a few. These components, when co-polymerized with different donor units such as those based on thiophene, or with electron-deficient units such as bithiazole, benzothiadiazole, benzobisthiadiazole and azine, afforded polymers with low-lying LUMO energy levels (well below −4.0 eV), remarkable ambient stability and field-effect mobilities larger than 0.5 cm2 V−1 s−1. The introduction of highly electron-withdrawing groups such as cyanine, chlorine and fluorine into the aforementioned building blocks has proven useful for improving the material performance.36 Fluorination, for example, can substantially lower the material LUMO energies and favour a sufficiently dense molecular packing, thus creating materials that are thermodynamically and kinetically resistant to trapping.
As we will see in the next section, one should be cautious of simply borrowing knowledge that has been gained for n-channel OFET materials and directly transferring it to OECTs. There are other effects that one needs to take into account in order to achieve stable, electrochemical n-type operation in an aqueous environment.
3.2 From OFETs to OECTs
In 2005, Panzer et al.48 reported one of the first OECTs based on n-type organic semiconductors. PDI, functionalized with dioctyl side chains (i.e., PTCDI-C8), was used as the transistor channel material, while a mixture of poly(ethyleneoxide) and lithium perchlorate was used as the electrolyte dielectric. Because of the limited ambient-stability of PTCDI-C8, the device was operated in vacuum. Two distinctive doping regions were identified depending on the applied gate voltage range: (i) an electrostatic doping regime occurring at VG < 2 V and (ii) an electrochemical doping regime for VG > 2 V. For the latter regime, diffusion of Li+ ions into the organic semiconductor film and subsequent conversion of all PTCDI-C8 molecules into anionic species was observed by vibrational spectroscopy.49 However, the transistors, operated in the electrochemical mode, suffered from large hysteresis and irreversible channel degradation. This was ascribed to partial trapping of Li+ in the PTCDI-C8 layer and chemical side reaction between the anionic PTCDI-C8 and contaminating water molecules contained in the electrolyte, which hampered any further application of PTCDI-C8-based OECTs in air and/or water-based media.Recently, Giovannitti at al.50 reported a material design strategy that allows for the development of n-type OECTs with stable operation in aqueous environments. The polymer backbone motif was based on the donor–acceptor poly([N,N′-bis(2-octyldodecyl)-naphthalene-1,4,5,8-bis(dicarboximide)-2,6-diyl]-alt-5,5′-(2,2′-bithiophene)) (P(NDI2OD-T2)) where the hydrophobic, branched 2-octyldodecyl (2OD) side chains were gradually exchanged for the more hydrophilic, linear heptakis(ethylene glycol) (HEG)-based side chains (Fig. 3A). P(NDI2OD-T2) (n = 0, Fig. 3A) is a notable example of n-type organic semiconductor.51 It represents a class of materials that can yield high electron mobility (0.2–0.8 cm2 V−1 s−1) in OFETs and is one of the most studied n-type polymers, frequently reported in recent literature. However, because of the hydrophobic nature of its 2OD side chains, only electrostatic charge accumulation at the semiconductor–electrolyte interface is typically reported for P(NDI2OD-T2).52 Electrochemical doping has not been observed within the potential window afforded by aqueous electrolytes, which is limited by the electrolysis of water (Fig. 3B). A series of random co-polymers was prepared with glycol side chain percentages ranging from 0 to 100 with respect to the alkyl side chains. Copolymers having a percentage of glycol side chain <50% showed a low degree of swelling (<10%) in 0.1 M NaCl aqueous solution. The swelling dramatically increased for glycol side chain content larger than 50% (>100% swelling for 100% glycol fraction), demonstrating a significant difference in the hydration behaviour depending on the nature of the side chains. A lower reduction potential as well as a lower hysteresis during electrochemical redox reactions was also observed for the co-polymers that showed higher degrees of swelling (Fig. 3C). As the co-polymers have identical backbones, the shift in the reduction potential was attributed to the difference in ion penetration into the bulk during electrochemical doping. This observation was corroborated by electrochemical impedance spectroscopy measurements showing that the polymers with glycol side chain percentage <10% had a lower capacitance compared to those with glycol side chain fraction >75% (Fig. 3D). The observed trend in capacitance suggests a gradual transition from an electrostatic accumulation of charges at the semiconductor–electrolyte interface for low glycol chain content, which is typical of EGOFETs based on P(NDI2OD-T2)53 and other hydrophobic semiconductors, to volumetric charging where ions are able to penetrate into the bulk of the co-polymer for glycol chain densities >75%. This volumetric doping of the semiconductor film resulted in a C* of about 200 F cm−3, and a thickness-normalized gm as high as 0.2 S cm−1, for the co-polymer with glycol fraction >90% (Fig. 3D). The large increase in C* was, however, compensated by a deterioration of the electronic charge transport properties. A significant drop in the electron mobility as well as an increase in the energetic disorder was in fact observed as the alkyl chains were replaced by the glycol side chains, suggesting the latter functionalization induces structural changes that contribute to disruption in electron mobility.
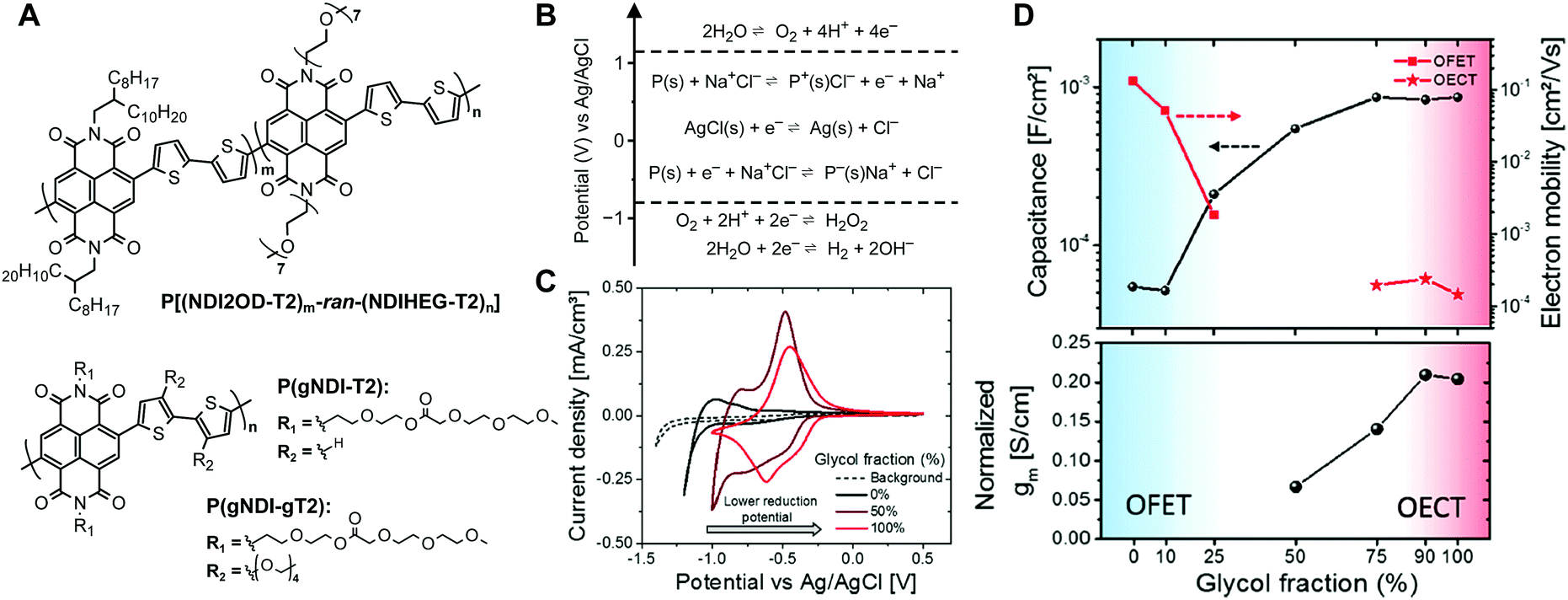 | ||
| Fig. 3 (A) Chemical structure of some NDI-based polymers for OECTs. (B) Electrochemical redox reactions of a polymer including the voltage limits of a stable operation in aqueous solution versus Ag/AgCl. Adapted with permission from ref. 54 Copyright The Authors. (C) Thin film cyclic voltammetry measurements of P[(NDI2OD-T2)m-ran-(NDIHEG-T2)n] with n = 0, 0.5 and 1 in 0.1 M NaCl aqueous solution (scan rate 100 mV s−1vs. Ag/AgCl). (D) Capacitance, electron mobility and normalized transconductance of P[(NDI2OD-T2)m-ran-(NDIHEG-T2)n] with n ranging from 0 to 1. Reprinted with permission from ref. 50 copyright 2018 American Chemical Society. | ||
The same group also showed that the choice of the hydrophilic side chain as well as of the donor co-monomer is highly important for the preparation of materials for n-type OECTs.54 Indeed, replacing linear glycol side chains with glycol-ester side chains, while keeping the same NDI-T2 backbone motif (Fig. 3A), resulted in electrochemical currents that were too low to be detected, although volumetric capacitance and field-effect mobilities remained unaffected (∼200 F cm−3 and 10−4 cm2 V−1 s−1, respectively). On the other hand, by replacing the bithiophene (T2) unit to an alkoxybithiophene (gT2) unit with increased donor strength (Fig. 3A), a two-fold increase in C* (∼400 F cm−3) was observed, yielding thickness-normalized gm of up to 1.0 S cm−1. The authors theorized that the increased electron affinity of NDI-gT2 along with less efficient packing of the polymer backbones led to more facile cation injection into the material and hence a reduced threshold voltage for the device.54
The nature of the donor co-monomer is known to influence the donor–acceptor character of the co-polymer affecting, to a great extent, the ability of the acceptor unit to stabilize more charges on the backbone. If these charges were highly mobile, this could augment the performance of n-type OECTs. However, NDI-based polymers such as those presented above typically suffer from only a modest electrical conductivity at high charge density (<0.01 S cm−1), due to the strong intrachain localization of the charge carriers, as well as to a limited tolerance of the flexible backbone for disorder induced by intercalation of ions, which is typical of this class of polymers.55 Reducing the donor–acceptor character as well as increasing the planarity of the polymer backbone has proven useful to enhance the electrical conductivity of NDI-based polymers by more than one order of magnitude.56 The use of highly planarized and rigid ladder-type polymers such as poly(benzobisimidazobenzophenanthroline) (BBL, Fig. 4A) has helped overcome this limitation, reaching a conductivity that is 1000× that of the backbone-distorted P(NDI2OD-T2).57 Our group recently demonstrated that ladder-type conducting polymers such as BBL can function as efficient electroactive materials for n-type OECTs.58 The transistors, fabricated by means of facile spray-coating techniques, function in accumulation mode in water with a channel-area-normalized gm that is about 2.5× higher than that of the best performing NDI-based OECTs of comparable thickness (Fig. 4B).54 The reason for the higher performance can be explained by considering the product μC*. Since no side chains are used to help solubilize the polymer backbone, BBL shows an impressive volumetric capacitance, over 900 F cm−3, at an offset voltage of −0.5 V vs. Ag/AgCl. Remarkably, this value exceeded that of the best NDI-based OECTs (∼400 F cm−3) by more than 2-fold,54 and was several orders of magnitude higher than the estimated value for NDI-based EGOFETs.53 In addition, because of the high electron conductivity and electron affinity, BBL-based OECTs operated at high currents in water and were stable during continuous pulse measurements over 1 h (Fig. 4C) as well as during three months of storage in air. However, the devices suffered from a slow response time due to the limited diffusion of ions throughout the BBL active layer. This limitation could perhaps be overcome by functionalizing the polymer backbone with chemical moieties that promote ion migration, although at the expense of the capacitance. Nevertheless, because side chains can ultimately influence both ions injection into the film and their effectiveness in doping the conjugated polymer,59 the disadvantage of a reduced capacitance could be mitigated by the improved response time. In addition to these advantages, side chain engineering could facilitate processing of the material. A complete report of the latest progress on fully conjugated ladder polymers and ways of improving their solubility can be found in recent reviews.60
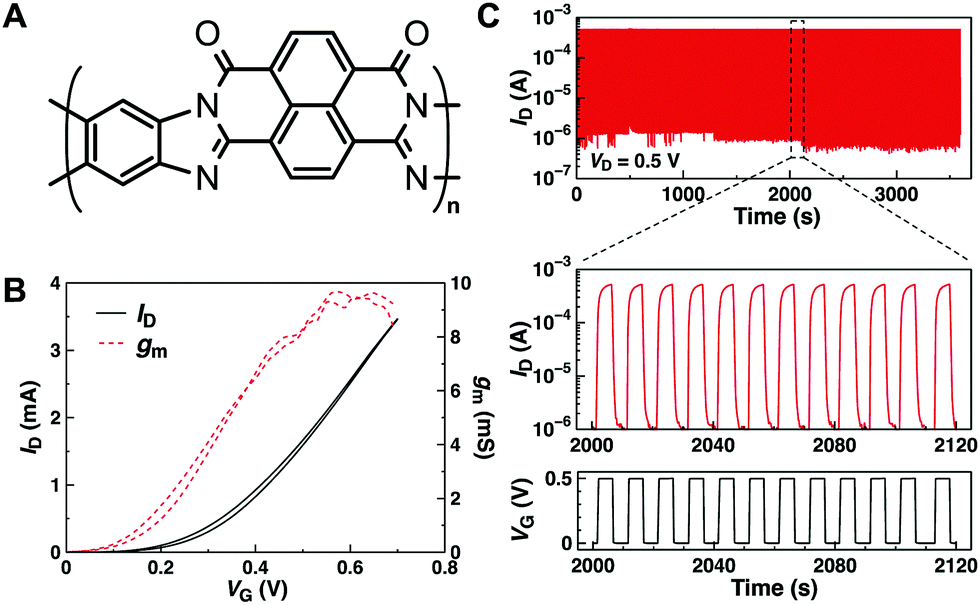 | ||
| Fig. 4 (A) Chemical structure of BBL. (B) n-Type transfer curve (black) and transconductance (red dotted) of a 180 nm-thick BBL OECT measured at VD = 0.6 V in 0.1 M NaCl. (C) Stability test of BBL OECT under sequential square wave gate voltage pulse for 1 h in water. Reprinted with permission from ref. 58 copyright The Authors. | ||
3.3 Applications: from complementary circuits to biosensors
Complementary circuits based entirely on OECT operation are currently at an early stage of development due to the scarcity of n-channel OECT materials with high electrical performance and ambient stability. Efficient n-channel conduction in organic materials is in fact crucial to the function of complementary organic circuits, where the combination of p- and n-channel transistors would enable greater circuit speeds, lower power dissipation, and more stable operation. Over the past decade, OECT circuits based on p-channel transistors have been realized for a variety of applications, ranging from pixel drivers for active-matrix electrochromic display to NAND/NOR gates,14 while the development of complementary circuits was limited to the use of solid-state OFETs.20 We recently reported one of the first example of complementary inverters based entirely on electrochemical operation by combining the best-performing BBL-based n-type OECTs with p-type OECTs based on the conjugated polyelectrolyte poly(3-carboxy-pentyl-thiophene) (P3CPT).58 The inverter operated at a supply voltage of 0.6 V, with relatively high gains (defined as ∂Vout/∂Vin) > 11, and large worst-case noise margin (NM) of 0.2 V (Fig. 5A and B). The NM was ≈67% of its maximum theoretical value of 0.3 V taking into account a supply voltage of 0.6 V. These values were larger than those reported for p-MOS inverters used in complex circuitry61 and among the best based on solution-processed organic CMOS-like inverters operating at such a low supply voltage (<0.6 V).52,53 Although still far from being technologically relevant in terms of speed, these devices represent a first example of CMOS-like inverters based on OECT operation that could find use as amplifying transducers to enhance the recorded signal of a biological event with low power consumption.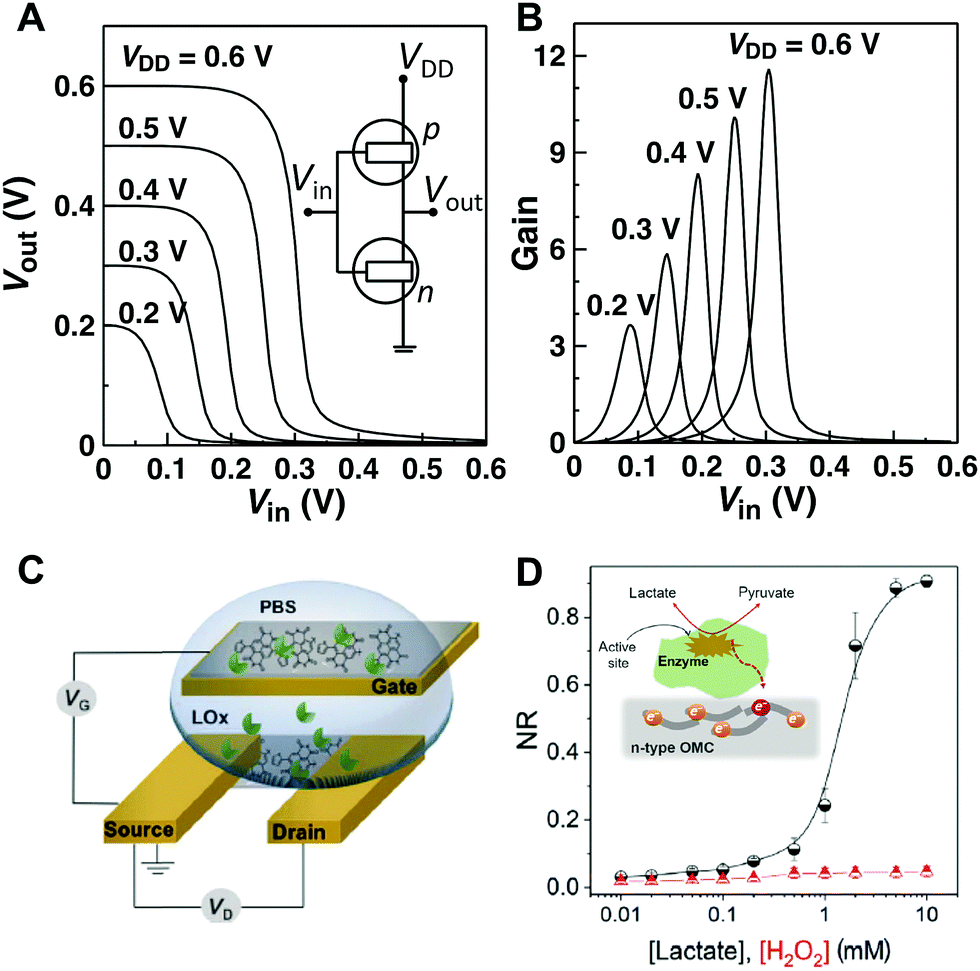 | ||
| Fig. 5 (A) Voltage characteristics and schematic representation of a complementary-like inverter based on BBL (n) and P3CPT (p) OECTs. (B) Corresponding voltage gains. Adapted with permission from ref. 58 Copyright The Authors. (C) Schematic of the OECT-based metabolite sensor. (D) Normalized response (NR) of the OECT to H2O2 (red) and to lactate in the presence of lactate oxidase (black). The inset shows proposed mechanism of lactate sensing based on the direct electron-transfer from the enzyme to the n-type channel. Adapted with permission from ref. 62 copyright The Authors. | ||
Recently, n-type OECTs have also been reported as an efficient platform for the detection of lactate, a significant metabolite in cellular metabolic pathways associated with critical health care conditions. The n-type conjugated polymer forming the OECT channel is capable of accepting electrons generated by the enzymatic reaction and acts as a series of redox centres capable of switching between the neutral and reduced states (Fig. 5C and D). This results in a fast, selective, and sensitive metabolite sensor, with obvious advantages over the traditional amperometric sensors in terms of amplification and design simplicity. The implications of this study go beyond the field of biosensors and open up an avenue for the development of enzyme-based electrocatalytic energy generation/storage devices.62
4. Conclusions and outlook
Although still at an early stage of their development, n-channel OECTs have attracted a great deal of attention, as they hold significant promise for improving the sophistication of the existing OECT-based technologies. The recent advances in n-type OECTs, as highlighted in this article, have benefitted from the understanding, gained over the years in the field of OFETs, of the factors limiting electron transport in n-channel materials, and the performance difference with the p-channel counterpart has substantially decreased. There are, however, several challenges that need to be addressed in order to push forward practical applications of n-channel OECT materials. For example, the best performing n-type OECT materials to date, such as p(gNDI-g2T) or BBL, have a μC* of ∼0.1–1 F cm−1 V−1 s−1, which is 1–2 orders of magnitude smaller than PEDOT:PSS-based p-type OECTs (∼80 F cm−1 V−1 s−1). This is not due to a reduced capability of the n-type material to store ionic charges, as evidence by a more than 20 times higher C* for BBL compared to PEDOT:PSS (∼900 vs. ∼40 F cm−3, respectively), but to a lower electron mobility (∼10−3vs. ∼2 cm2 V−1 s−1 for BBL and PEDOT:PSS, respectively). The use of conjugated polymers with planar, torsion-free backbones have emerged as a promising strategy to reduce intrinsic conformational and energetic disorder and to boost charge carrier mobility.63 To this end, fully conjugated ladder-type polymers or polymers where double bonds lock-in a rigid backbone conformation64 hold promises for the development of conducting channel materials with efficient and air stable electron transport. In addition, high carrier mobilities should be achievable through a simultaneous improvement of the local aggregation and interconnectivity of domains. Thus, research should focus on the interchain coupling in the aggregates and/or on the design of molecules that afford delocalization.65Hence, a specific challenge to materials chemists would be then to design solution-processable organic semiconductors that have high stability in air and aqueous electrolyte media, high electrochemical stability within the working potential range of the electrolyte–solvent system, and be chemically and physically tolerant to disorder at high doping levels during ion penetration into the channel volume, by allowing more efficient intra- and intermolecular charge transport pathways, while maintaining high electron mobility. Despite these challenges, we believe that the development of n-type OECTs has a bright future, especially considering that the vast amount of previous work on both p-type OECTs and n-channel OFETs may provide useful insights to tackle these challenges, helping to advance this field in the direction of practical technologies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Swedish Research Council (2016-03979), the Swedish Governmental Agency for Innovation Systems – VINNOVA (2015-04859), the Swedish Foundation for Strategic Research (SE13-0045 and RIT15-0119), and the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO-Mat-LiU # 2009-00971).References
- D. T. Simon, E. O. Gabrielsson, K. Tybrandt and M. Berggren, Chem. Rev., 2016, 116, 13009–13041 CrossRef PubMed.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef.
- P. Lin, F. Yan, J. J. Yu, H. L. W. Chan and M. Yang, Adv. Mater., 2010, 22, 3655–3660 CrossRef PubMed.
- P. Lin, X. T. Luo, I. M. Hsing and F. Yan, Adv. Mater., 2011, 23, 4035–4040 CrossRef PubMed.
- D. Khodagholy, V. F. Curto, K. J. Fraser, M. Gurfinkel, R. Byrne, D. Diamond, G. G. Malliaras, F. Benito-Lopez and R. M. Owens, J. Mater. Chem., 2012, 22, 4440–4443 RSC.
- L. H. Jimison, S. A. Tria, D. Khodagholy, M. Gurfinkel, E. Lanzarini, A. Hama, G. G. Malliaras and R. M. Owens, Adv. Mater., 2012, 24, 5919–5923 CrossRef PubMed.
- G. Tarabella, P. D’Angelo, A. Cifarelli, A. Dimonte, A. Romeo, T. Berzina, V. Erokhin and S. Iannotta, Chem. Sci., 2015, 6, 2859–2868 RSC.
- F. Hempel, J. K. Y. Law, T. C. Nguyen, W. Munief, X. L. Lu, V. Pachauri, A. Susloparova, X. T. Vu and S. Ingebrandt, Biosens. Bioelectron., 2017, 93, 132–138 CrossRef PubMed.
- O. Salyk, J. Vitecek, L. Omasta, E. Safarikova, S. Stritesky, M. Vala and M. Weiter, Appl. Sci., 2017, 7, 998–1007 CrossRef.
- A. Campana, T. Cramer, D. T. Simon, M. Berggren and F. Biscarini, Adv. Mater., 2014, 26, 3874–3878 CrossRef PubMed.
- P. Leleux, C. Johnson, X. Strakosas, J. Rivnay, T. Herve, R. M. Owens and G. G. Malliaras, Adv. Healthcare Mater., 2014, 3, 1377–1380 CrossRef PubMed.
- Y. van de Burgt, E. Lubberman, E. J. Fuller, S. T. Keene, G. C. Faria, S. Agarwal, M. J. Marinella, A. Alec Talin and A. Salleo, Nat. Mater., 2017, 16, 414–418 CrossRef PubMed.
- P. Gkoupidenis, D. A. Koutsouras and G. G. Malliaras, Nat. Commun., 2017, 8, 15448 CrossRef PubMed.
- D. Nilsson, N. Robinson, M. Berggren and R. Forchheimer, Adv. Mater., 2005, 17, 353–358 CrossRef.
- P. A. Ersman, D. Westerberg, D. Y. Tu, M. Nilsson, J. Ahlin, A. Eveborn, A. Lagerlof, D. Nilsson, M. Sandberg, P. Norberg, M. Berggren, R. Forchheimer and G. Gustafsson, Flexible Printed Electron., 2017, 2, 045008 CrossRef.
- E. Zeglio, M. Vagin, C. Musumeci, F. N. Ajjan, R. Gabrielsson, X. T. Trinh, N. T. Son, A. Maziz, N. Solin and O. Inganäs, Chem. Mater., 2015, 27, 6385–6393 CrossRef.
- S. Fabiano, N. Sani, J. Kawahara, L. Kergoat, J. Nissa, I. Engquist, X. Crispin and M. Berggren, Sci. Adv., 2017, 3, e1700345 CrossRef PubMed.
- M. Hamedi, R. Forchheimer and O. Inganas, Nat. Mater., 2007, 6, 357–362 CrossRef PubMed.
- A. Facchetti, Mater. Today, 2007, 10, 28–37 CrossRef.
- K. J. Baeg, M. Caironi and Y. Y. Noh, Adv. Mater., 2013, 25, 4210–4244 CrossRef PubMed.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef PubMed.
- H. Koezuka, A. Tsumura and T. Ando, Synth. Met., 1987, 18, 699–704 CrossRef.
- S. H. Kim, K. Hong, W. Xie, K. H. Lee, S. Zhang, T. P. Lodge and D. C. Frisbie, Adv. Mater., 2013, 25, 1822–1846 CrossRef PubMed.
- D. Zhao, S. Fabiano, M. Berggren and X. Crispin, Nat. Commun., 2017, 8, 14214 CrossRef PubMed.
- Y. Xia, W. Zhang, M. Ha, J. H. Cho, M. J. Renn, C. H. Kim and C. D. Frisbie, Adv. Funct. Mater., 2010, 20, 587–594 CrossRef.
- J. Rivnay, P. Leleux, M. Ferro, M. Sessolo, A. Williamson, D. A. Koutsouras, D. Khodagholy, M. Ramuz, X. Strakosas, R. M. Owens, C. Benar, J.-M. Badier, C. Bernard and G. G. Malliaras, Sci. Adv., 2015, 1, e1400251 Search PubMed.
- O. Larsson, A. Laiho, W. Schmickler, M. Berggren and X. Crispin, Adv. Mater., 2011, 23, 4764–4769 CrossRef PubMed.
- D. Khodagholy, J. Rivnay, M. Sessolo, M. Gurfinkel, P. Leleux, L. H. Jimison, E. Stavrinidou, T. Herve, S. Sanaur, R. M. Owens and G. G. Malliaras, Nat. Commun., 2013, 4, 2133 CrossRef PubMed.
- A. Laiho, L. Herlogsson, R. Forchheimer, X. Crispin and M. Berggren, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15069–15073 CrossRef PubMed.
- S. G. Bucella, A. Luzio, E. Gann, L. Thomsen, C. R. McNeill, G. Pace, A. Perinot, Z. Chen, A. Facchetti and M. Caironi, Nat. Commun., 2015, 6, 8394 CrossRef PubMed.
- D. A. Bernards and G. G. Malliaras, Adv. Funct. Mater., 2007, 17, 3538–3544 CrossRef.
- H. L. Dong, X. L. Fu, J. Liu, Z. R. Wang and W. P. Hu, Adv. Mater., 2013, 25, 6158–6182 CrossRef PubMed.
- S. Inal, G. G. Malliaras and J. Rivnay, Nat. Commun., 2017, 8, 1767 CrossRef PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef.
- D. M. de Leeuw, M. M. J. Simenon, A. R. Brown and R. E. F. Einerhand, Synth. Met., 1997, 87, 53–59 CrossRef.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef PubMed.
- D. Natali and M. Caironi, Adv. Mater., 2012, 24, 1357–1387 CrossRef PubMed.
- H. Usta, A. Facchetti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501–510 CrossRef PubMed.
- X. Zhao and X. Zhan, Chem. Soc. Rev., 2011, 40, 3728–3743 RSC.
- S. Holliday, J. E. Donaghey and I. McCulloch, Chem. Mater., 2014, 26, 647–663 CrossRef.
- J. T. E. Quinn, J. Zhu, X. Li, J. Wang and Y. Li, J. Mater. Chem. C, 2017, 5, 8654–8681 RSC.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef PubMed.
- Z. H. Chen, Y. Zheng, H. Yan and A. Facchetti, J. Am. Chem. Soc., 2009, 131, 8–9 CrossRef PubMed.
- M. Sommer, J. Mater. Chem. C, 2014, 2, 3088–3098 RSC.
- B. Nielsen Christian, M. Turbiez and I. McCulloch, Adv. Mater., 2012, 25, 1859–1880 CrossRef PubMed.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2014, 26, 664–678 CrossRef.
- Z. Yan, B. Sun and Y. Li, Chem. Commun., 2013, 49, 3790–3792 RSC.
- M. J. Panzer and C. D. Frisbie, J. Am. Chem. Soc., 2005, 127, 6960–6961 CrossRef PubMed.
- L. G. Kaake, Y. Zou, M. J. Panzer, C. D. Frisbie and X. Y. Zhu, J. Am. Chem. Soc., 2007, 129, 7824–7830 CrossRef PubMed.
- A. Giovannitti, I. P. Maria, D. Hanifi, M. J. Donahue, D. Bryant, K. J. Barth, B. E. Makdah, A. Savva, D. Moia, M. Zetek, P. R. F. Barnes, O. G. Reid, S. Inal, G. Rumbles, G. G. Malliaras, J. Nelson, J. Rivnay and I. McCulloch, Chem. Mater., 2018, 30, 2945–2953 CrossRef PubMed.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef PubMed.
- L. Herlogsson, X. Crispin, S. Tierney and M. Berggren, Adv. Mater., 2011, 23, 4684–4689 CrossRef PubMed.
- R. Porrazzo, A. Luzio, S. Bellani, G. E. Bonacchini, Y.-Y. Noh, Y.-H. Kim, G. Lanzani, M. R. Antognazza and M. Caironi, ACS Omega, 2017, 2, 1–10 CrossRef PubMed.
- A. Giovannitti, C. B. Nielsen, D.-T. Sbircea, S. Inal, M. Donahue, M. R. Niazi, D. A. Hanifi, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, Nat. Commun., 2016, 7, 13066 CrossRef PubMed.
- B. D. Naab, X. Gu, T. Kurosawa, J. W. F. To, A. Salleo and Z. Bao, Adv. Electron. Mater., 2016, 2, 1600004 CrossRef.
- S. Wang, H. Sun, T. Erdmann, G. Wang, D. Fazzi, U. Lappan, Y. Puttisong, Z. Chen, M. Berggren, X. Crispin, A. Kiriy, B. Voit, T. J. Marks, S. Fabiano and A. Facchetti, Adv. Mater., 2018, 30, 1801898 CrossRef PubMed.
- S. Wang, H. Sun, U. Ail, M. Vagin, P. O. Å. Persson, J. W. Andreasen, W. Thiel, M. Berggren, X. Crispin, D. Fazzi and S. Fabiano, Adv. Mater., 2016, 28, 10764–10771 CrossRef PubMed.
- H. Sun, M. Vagin, S. Wang, X. Crispin, R. Forchheimer, M. Berggren and S. Fabiano, Adv. Mater., 2018, 30, 1704916 CrossRef PubMed.
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Müller, ACS Energy Lett., 2018, 3, 278–285 CrossRef PubMed.
- J. Lee, A. J. Kalin, T. Yuan, M. Al-Hashimi and L. Fang, Chem. Sci., 2017, 8, 2503–2521 RSC.
- J. H. Cho, J. Lee, Y. Xia, B. Kim, Y. Y. He, M. J. Renn, T. P. Lodge and C. D. Frisbie, Nat. Mater., 2008, 7, 900–906 CrossRef PubMed.
- A. M. Pappa, D. Ohayon, A. Giovannitti, I. P. Maria, A. Savva, I. Uguz, J. Rivnay, I. McCulloch, R. M. Owens and S. Inal, Sci. Adv., 2018, 4, eaat0911 CrossRef PubMed.
- D. Venkateshvaran, M. Nikolka, A. Sadhanala, V. Lemaur, M. Zelazny, M. Kepa, M. Hurhangee, A. J. Kronemeijer, V. Pecunia, I. Nasrallah, I. Romanov, K. Broch, I. McCulloch, D. Emin, Y. Olivier, J. Cornil, D. Beljonne and H. Sirringhaus, Nature, 2014, 515, 384–388 CrossRef PubMed.
- A. Onwubiko, W. Yue, C. Jellett, M. Xiao, H.-Y. Chen, M. K. Ravva, D. A. Hanifi, A.-C. Knall, B. Purushothaman, M. Nikolka, J.-C. Flores, A. Salleo, J.-L. Bredas, H. Sirringhaus, P. Hayoz and I. McCulloch, Nat. Commun., 2018, 9, 416 CrossRef PubMed.
- S. Wang, S. Fabiano, S. Himmelberger, S. Puzinas, X. Crispin, A. Salleo and M. Berggren, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10599–10604 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |