
Bifuran-imide: a stable furan building unit for organic electronics†
Ori
Gidron  *a
*a
*a
Abstract
Oligo- and polyfurans hold many advantages over their thiophene analogues, yet their low environmental stability greatly limits their applications. In this work, we introduce a new building unit, bifuran-imide (BFI), which displays many of the advantageous properties observed for oligofurans, such as strong fluorescence, high solubility, significant quinoid character, and planarity, while also being significantly more stable (both thermally and photochemically) than parent oligofurans. Oligomers and polymers containing BFI show good solid state packing, strong fluorescence, and solubility, which makes them ideal candidates for active materials in organic electronics.
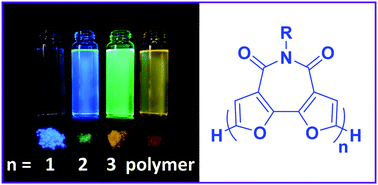
- This article is part of the themed collection: Journal of Materials Chemistry C Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

