
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWeak thermal quenching of the luminescence in the Ca3Sc2Si3O12:Ce3+ garnet phosphor†
Suchinder K.
Sharma‡
 a,
Yuan-Chih
Lin
a,
Irene
Carrasco§
b,
Tobias
Tingberg
c,
Marco
Bettinelli
b and
Maths
Karlsson
*a
a,
Yuan-Chih
Lin
a,
Irene
Carrasco§
b,
Tobias
Tingberg
c,
Marco
Bettinelli
b and
Maths
Karlsson
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-412 96 Göteborg, Sweden. E-mail: maths.karlsson@chalmers.se; Tel: +46 31 772 6770
bLuminescent Materials Laboratory, Department of Biotechnology, University of Verona, and INSTM, UdR Verona, Strada Le Grazie 15, 371 34 Verona, Italy
cDepartment of Microtechnology and Nanoscience, Chalmers University of Technology, 412 96 Göteborg, Sweden
First published on 26th July 2018
Abstract
We report results of the luminescence properties of the three garnet type phosphors Ce3+-doped Ca3Sc2Si3O12 (CSSO:Ce3+), Sr3Y2Ge3O12 (SYG:Ce3+) and Y3Al5O12 (YAG:Ce3+), investigated using optical spectroscopy techniques and vacuum referred binding energy (VRBE) diagram analysis. By monitoring the temperature dependence of the luminescence decay time we establish an excellent, intrinsic, thermal stability of luminescence in CSSO:Ce3+, with a nearly constant decay time (≈60 ns) up to, at least, T = 860 K. In comparison, SYG:Ce3+ and YAG:Ce3+ exhibit a significant reduction of the luminescence decay time upon heating, starting at around T = 280 K and T = 550 K, respectively, suggesting a lower internal thermal stability of luminescence in these two garnet phosphors. These findings are supported by the energy separation between the Ce3+ 5d1 level and the conduction band (CB) of the respective hosts, which are found at 1.36 eV (CSSO:Ce3+), 0.45 eV (SYG:Ce3+), and 1.17 eV (YAG:Ce3+), respectively, as predicted by their VRBE diagrams. The performance of CSSO:Ce3+ was evaluated by applying the phosphor on a blue InGaN LED. The system shows a luminous efficacy of optical radiation of 243 lm W−1 and a linear response with increasing applied voltage, suggesting it is a highly promising phosphor for future technological applications, particularly at high temperature operating environments.
1 Introduction
Phosphor-converted white-light emitting diodes (pc-WLEDs) based on blue InGaN LEDs offer many advantages compared to traditional light sources due to their high efficiency, tuneable color, and long lifetimes.1–6 The phosphors consist of a crystalline host lattice containing a small amount of luminescent ions, which convert the blue light to longer wavelengths via electronic transitions. Garnet crystals, of general formula A3B2C3O12, where B and C may be the same or different atoms and as characterized by a 160-atom body-centered cubic unit cell with a lattice constant L [Fig. 1(a)], are widely considered to be one of the most important families of hosts for luminescent ions, especially for trivalent lanthanide ions such as Ce3+. The A, B, and C atoms are (8-fold) dodecahedrally, (6-fold) octahedrally, and (4-fold) tetrahedrally coordinated to O atoms that are shared between neighboring cation–oxygen polyhedra.7 The choice of cations provides a means to tune the local coordination environment of the luminescent ions and thus important materials properties of relevance for luminescence, such as the centroid shift (εc) and crystal field splitting (Δ and ΔE5d1–2) of the luminescent ions;7–12 see Fig. 1(b) for a schematic illustration of the effect on the electronic energy levels of a free Ce3+ ion when inserted in a host.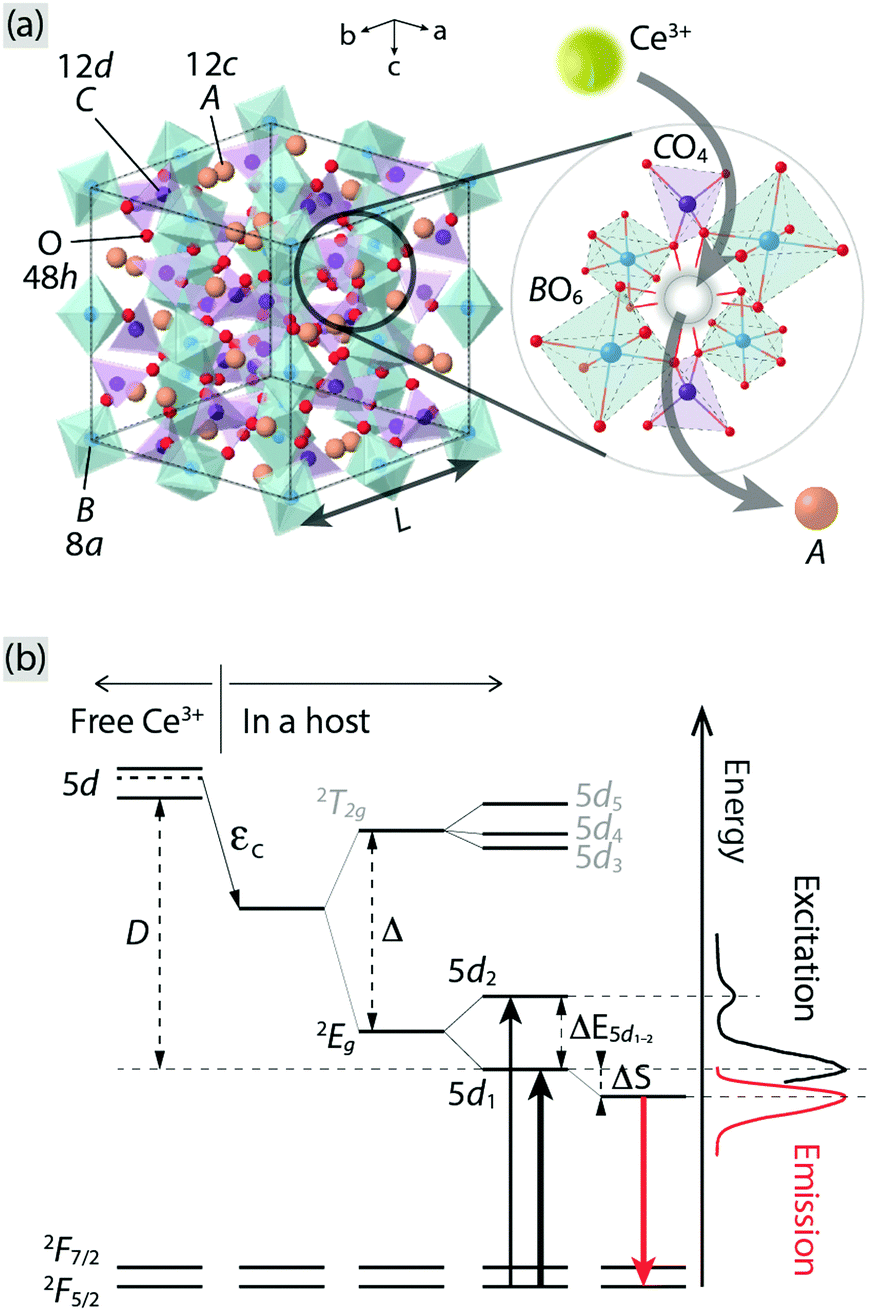 | ||
| Fig. 1 (a) Illustration of an unit cell of a A3B2C3O12 garnet crystal. The A, B, C, and O atoms occupy Wyckoff positions 12c, 8a, 12d, and 48h, respectively. The image to the right shows a close-up of the local coordination of an A atom, which is oriented in a way that the neighboring polyhedra can be clearly seen, showing the substitution of Ce3+ for an A ion. (b) A schematic energy level diagram for a free Ce3+ and when it is embedded in a host, in relation to the PL spectra of Ce3+-doped Y3Al5O12 as an example (cf.Fig. 2). εc: centroid shift, Δ: crystal field splitting due to a cubal coordination, ΔE5d1–2: crystal field splitting due to tetragonally distorted coordination, ΔS: Stokes shift, D: red-shift of the lowest 5d level of Ce3+, 2T2g: triply degenerate level of the upper 5d levels of Ce3+, 2Eg: doubly degenerate level of the lower 5d levels of Ce3+, and 2F5/2 and 2F7/2: the split 4f levels of Ce3+ due to the spin–orbit coupling. | ||
The best known garnet is by far yttrium aluminium garnet (Y3Al5O12, YAG) that, when doped with Ce3+ (YAG:Ce3+), emits light in the green-yellow range due to inter-configurational 5d–4f transitions and constitutes an important phosphor for use in pc-WLEDs.13 The YAG structure (L = 12.01 Å) is built up of AlO6 octahedra and AlO4 tetrahedra, and YO8 dodecahedra, and the Ce3+ substitutes for Y3+ due to matching ionic charge and radii (cf. 1.143 Å for Ce3+ and 1.019 Å for Y3+, in 8-fold coordination).14–16 Two novel, green-emitting, analogous garnet systems to YAG:Ce3+, which can shed further light on the luminescent properties of garnet phosphors, are Ce3+-doped Ca3Sc2Si3O12 (CSSO:Ce3+) and Ce3+-doped Sr3Y2Ge3O12 (SYG:Ce3+). CSSO and SYG are isostructural with L = 12.25 Å and L = 13.08 Å, respectively.17,18 On the basis of considerations of the ionic radii, Ce3+ is expected to substitute for Ca2+/Sr2+ (cf. 1.143 Å for Ce3+ and 1.12 Å for Ca2+ and 1.26 Å for Sr2+, in 8-fold coordination),15 but, due to the charge mismatch, two Ce3+ ions are expected to substitute for three Ca2+/Sr2+ ions to maintain charge neutrality. Simultaneously, cation vacancies are formed as a charge compensating effect.
A critical challenge in the development of pc-WLEDs is to develop phosphors with high luminance and high thermal stability.19–21 In the literature available on YAG:Ce3+, CSSO:Ce3+, and SYG:Ce3+, the thermal stability of luminescence of YAG:Ce3+ has been studied extensively. YAG:Ce3+ exhibits a 20% loss of the room temperature intensity at T = 500 K and more than 50% at T = 700 K, depending on Ce3+ dopant concentration and excitation wavelength.22,23 The lowering of the emission intensity as a function of increasing temperature has been attributed to a combination of a change in the 4f–5d absorption strength, as well as (intrinsic) thermal quenching of luminescence, as inferred by a decrease in the luminescence decay time of the Ce3+ emission.22 For CSSO:Ce3+, the decrease in emission intensity with increasing temperature is less pronounced than for YAG:Ce3+ and has been also attributed to a change of the 4f–5d absorption strength, partly related to absorption by impurity phases (β-Ca2SiO4, CaSiO3, CeO2), whereas the luminescence decay time is nearly constant (≈71 ns) up to T = 700 K.24 The higher intrinsic thermal stability with respect to luminescence of CSSO:Ce3+ as compared to YAG:Ce3+ not only makes it a promising phosphor for application in pc-WLEDs, but it is also highly interesting from a more fundamental point of view. For the structurally similar phosphor SYG:Ce3+, there is a lack of information as the thermal stability of luminescence has not been investigated, yet.
The aim of the present work is to compare and understand the emission and excitation characteristics, especially in regard to the intrinsic thermal stability, of SYG:Ce3+, CSSO:Ce3+, and YAG:Ce3+ upon excitation at 420–470 nm, thus mimicking the experimental conditions for a pc-WLED using a blue LED. With a view toward practical applications, we also evaluate the performance of CSSO:Ce3+ phosphor in a pc-WLED prototype. Combined analyses of the emission and excitation spectra, the temperature dependence of the luminescence decay time of Ce3+ in SYG:Ce3+, CSSO:Ce3+, and YAG:Ce3+, and the vacuum referred binding energy (VRBE) diagram for Ce3+ in the respective host lattices, suggest that the separation between the host CB and the Ce3+ 5d1 level plays the most critical role in determining the thermal stability of the three garnet type phosphors.
2 Experimental
2.1 Sample preparation
CSSO:Ce3+ was prepared through solid state sintering, by mixing stoichiometric amounts of CaCO3 (Aldrich, ≥99% purity), Sc2O3 (Strem, 99.99% purity), SiO2 (Aldrich, ≥99.995% purity), and Ce(NO3)3·6H2O (Aldrich, 99.99% purity), followed by three repetitive heat treatments for 3 h at 1450 °C, with intermediate grindings and compacting of pellets under ambient conditions. SYG:Ce3+ was prepared by solid state sintering, by mixing stoichiometric amounts of AR grade SrCO3 (Aldrich, ≥99% purity), Y2O3 (Aldrich, 99.99% purity), GeO2 (Aldrich, 99.998% purity), and Ce(NO3)3·6H2O (Aldrich, 99.99% purity), followed by three repetitive heat treatments at 1250 °C (24 h), with intermediate grindings and compacting of pellets. YAG:Ce3+ was prepared by co-precipitation. Aqueous solutions of Y(NO3)3·6H2O (Strem, 99.999% purity), Ce(NO3)3·6H2O (Aldrich, 99.99% purity), and NH4Al(SO4)2·12H2O (Aldrich, >99% purity) were prepared from AR reagents. The mixed nitrate and sulfate solution was slowly added to a 3 M solution of ammonium hydroxide, keeping the pH > 10. The precipitate was washed with diluted ammonia and ethanol, filtered and dried overnight at 90 °C to obtain the precursor powder, which was subsequently annealed at 1100 °C for 2 h. The as-prepared samples were phase pure with no detectable traces of impurities as evaluated from powder X-ray diffraction patterns, collected on a Thermo ARL X'TRA powder diffractometer, operating in the Bragg–Brentano geometry and equipped with a Cu-anode X-ray source (Kα, λ = 1.5418 Å), using a Peltier Si (Li) cooled solid state detector. The patterns were collected with a scan rate of 0.03° s−1 in the 2θ range 5–90°.2.2 Photoluminescence spectroscopy
The PL excitation and emission spectra were measured using two different setups. Room temperature PL spectra were measured at the University of Verona, Italy, using a Fluorolog 3 spectrofluorometer (Horiba-Jobin Yvon), equipped with a Xe lamp, a double excitation monochromator, a single emission monochromator (mod. HR320) and a photomultiplier in photon counting mode for the detection of the emitted signal. Variable temperature emission spectra and luminescence decay curves were measured at Chalmers University of Technology, Sweden, over the temperature range T = 80–860 K. The temperature was controlled using a Linkam THMS 600 heating stage. The excitation was achieved using a 454 nm laser (DeltaDiode DD-450L) connected to a DD-Ci picosecond diode controller from Horiba Scientific. Due to a very weak and relatively broad emission band (420–490 nm) from the laser, which overlaps partly with the shorter-wavelength part of the emission bands of the studied phosphors, the laser light was further monochromatized by the use of a monochromator (300–800 nm Manual Mini-Chrom, Edmund Optics). Upon the pulsed excitation with a repetition rate of 500 kHz, luminescence decay curves were then detected using a H10721-20 photosensor module (Hamamatsu) containing a photomultiplier and a high-voltage power supply circuit in the 230–920 nm wavelength range, which was coupled to a 500 nm longpass filter (A10033-62, Hamamatsu). The detected decay curves were recorded on an Agilent DSO-X 2022A oscilloscope. The emission spectra were measured as the samples were excited by the 454 nm pulsed laser with a repetition rate of 100 MHz, by using an Ocean Optics USB2000+ UV-Vis spectrometer coupled to an optical fiber with a 455 nm longpass colored glass filter (FGL455, Thorlabs) attached in front of the fiber inlet. Reference spectra were measured using the same setting as described above, but without the blue light irradiating on the samples, and subtracted from the spectra of the samples.2.3 Phosphor-converted WLED prototype
In order to evaluate the actual performance of CSSO:Ce3+ phosphor in a pc-WLED device, we integrated this phosphor onto a commercial 420 nm emitting blue LED chip, using the novel pc-WLED design, as developed by Bin Im et al.25 The phosphor was molded with silicon resin, put on a cap that was placed onto the LED, and left to solidify for a period over 24 h. A small air gap (≈5 mm; refractive index n = 1) between the LED chip and phosphor cap (n ≈ 1.7) reduces the amount of light that is reflected back into the chip, due to the differences in refractive indices of the various layers. Further, the phosphor layer is insulated from the heat as generated by the LED chip, thus reducing a loss of emission intensity due to thermal quenching effects.The spectral output of the pc-WLED prototype was monitored on a OL 770 multi-channel spectroradiometer from Optronic Laboratories at Chalmers University of Technology, Göteborg. The input current was varied using a DC power supply from BK Precision.
3 Results
3.1 Photoluminescence spectra
The room temperature PL excitation and emission spectra of CSSO:Ce3+, SYG:Ce3+, and YAG:Ce3+ are shown in Fig. 2. The excitation spectra (black color) were measured at a fixed emission wavelength of 504 nm (CSSO:Ce3+), 513 nm (SYG:Ce3+), and 525 nm (YAG:Ce3+), respectively. The spectra possess excitation maxima at approximately 440 nm (CSSO:Ce3+), 427 nm (SYG:Ce3+), and 470 nm (YAG:Ce3+), with a full width at half maximum (FWHM) of 73 nm, 53 nm, and 72 nm, respectively, for the three materials. The excitation bands correspond to Ce3+ 4f → 5d1 transitions (λ5d1) in the respective hosts. Higher-energy bands peaking at approximately 285 nm (CSSO:Ce3+), 297 nm (SYG:Ce3+), and 340 nm (YAG:Ce3+), which correspond to Ce3+ 4f → 5d2 transitions (λ5d2), can be also observed. The energy difference (ΔE5d1–2) between the band maxima of the excitation transitions to the 5d2 and 5d1 levels is 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 360 cm−1, 10251 cm−1, and 8135 cm−1, for CSSO:Ce3+, SYG:Ce3+, and YAG:Ce3+, respectively, and can be used as a measure for the strength of “tetragonal” crystal-field splitting of the 8-fold coordinated Ce3+ ions.11,26 The ΔE5d1–2 values are in good agreement with the literature: for CSSO:Ce3+, the value (12360 cm−1) is very similar to the one reported for Ce3+-doped Lu2CaMg2Si3O12 (a similar Ce3+-doped silicate garnet) with ΔE5d1–2 = 11000–12000 cm−1.27 In the case of YAG:Ce3+, it is also comparable with previously reported ones, e.g. 8135 cm−1 (this work) vs. 7600 cm−1 by Setlur et al.27 The spectroscopic data of λ5d1, λ5d2, and ΔE5d1–2 are summarized in Table 1.
360 cm−1, 10251 cm−1, and 8135 cm−1, for CSSO:Ce3+, SYG:Ce3+, and YAG:Ce3+, respectively, and can be used as a measure for the strength of “tetragonal” crystal-field splitting of the 8-fold coordinated Ce3+ ions.11,26 The ΔE5d1–2 values are in good agreement with the literature: for CSSO:Ce3+, the value (12360 cm−1) is very similar to the one reported for Ce3+-doped Lu2CaMg2Si3O12 (a similar Ce3+-doped silicate garnet) with ΔE5d1–2 = 11000–12000 cm−1.27 In the case of YAG:Ce3+, it is also comparable with previously reported ones, e.g. 8135 cm−1 (this work) vs. 7600 cm−1 by Setlur et al.27 The spectroscopic data of λ5d1, λ5d2, and ΔE5d1–2 are summarized in Table 1.
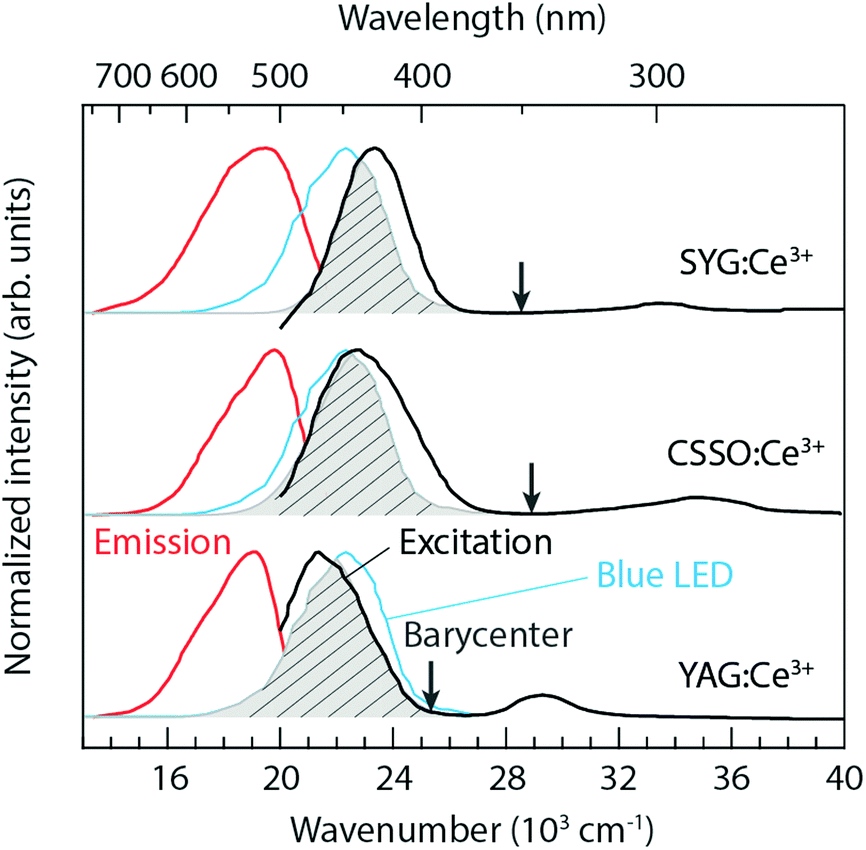 | ||
| Fig. 2 PL excitation (black color) and emission (red color) spectra for SYG:Ce3+, CSSO:Ce3+, and YAG:Ce3+, as measured at room temperature. The arrows indicate the 2Eg barycenter of the Ce3+ 5d levels. The emission spectrum (blue color) corresponding to an InGaN blue LED is included for comparison. The PL (4f → 5d1 and 5d1 → 4f) spectra of the three materials are normalized to the emission maximum of the blue LED. The spectral overlaps between the materials (excitation) and the blue LED (emission) are marked by the shaded grey areas. | ||
| Sample | λ 5d2 (nm) | λ 5d1 (nm) | λ 4f (nm) | ΔE5d1–2 (cm−1) | ΔS (cm−1) | (x, y) | CCT (K) | T 80% (K) | E 5d1-CB /Eac (eV) |
|---|---|---|---|---|---|---|---|---|---|
| a T 80% is the temperature at which the luminescence decay time has decreased to 80% with respect to the value at low temperature (here 80 K), i.e. with respect to that of the “intrinsic” decay time also known as lifetime. b Estimated from the VRBE diagrams (Fig. 6). c E a is obtained from the fits of the temperature dependent luminescence decay time [Fig. 5(d)] to a single-barrier quenching model. d Estimated from E5d1-CB of CSSO:Ce3+, using the relation between E5d1-CB and Ea of SYG:Ce3+ and YAG:Ce3+, respectively. | |||||||||
| SYG:Ce3+ | 297 | 427 | 513 | 10251 |
3926 | (0.314, 0.509) | 5911 | 280 | 0.45/0.33 |
| CSSO:Ce3+ | 285 | 440 | 504 | 12360 |
2886 | (0.273, 0.542) | 6749 | >860 | 1.36/0.5–1.0d |
| YAG:Ce3+ | 340 | 470 | 525 | 8135 | 2229 | (0.355, 0.576) | 5130 | 550 | 1.17/0.45 |
The Ce3+ 4f → 5d1 excitation band of YAG:Ce3+ is well located within the InGaN blue emission. The spectral overlap between the excitation band of YAG:Ce3+ and the emission band of an InGaN blue LED is about 85% under the circumstance that the maxima of the 4f → 5d1 excitation bands are normalized to the emission maximum of the blue LED, see the shaded grey area in Fig. 2. This suggests a high QE of the conversion of blue light into green-yellow light, which is an important criterion for most practical applications.28 In comparison, the spectral overlaps are around 83% and 58%, for CSSO:Ce3+ and SYG:Ce3+, respectively, suggesting a lower QE for these materials.
The emission spectra (red color) were measured by fixing the excitation wavelength at 440 nm (CSSO:Ce3+), 427 nm (SYG:Ce3+), and 470 nm (YAG:Ce3+). The spectral maxima of the 5d1 → 4f emission taking place after a relaxation (Stokes shift, ΔS) from higher to lower vibrational states of the Ce3+ 5d1 level, as denoted by λ4f in Table 1, are shifted with respect to each other, with the spectrum peaking at 504 nm for CSSO:Ce3+ (ΔS = 2886 cm−1), at around 513 nm for SYG:Ce3+ (ΔS = 3926 cm−1), and at around 525 nm for YAG:Ce3+ (ΔS = 2229 cm−1). A trend of shifting the emission color from green for CSSO:Ce3+ and SYG:Ce3+ to greenish-yellow light for YAG:Ce3+ is evident from the CIE 1931 diagram (Fig. 3). The corresponding color coordinates (x and y) are shown in Table 1. Included in Table 1 are also the correlated color temperatures (CCTs) at T = 300 K, as determined from the respective color coordinates.
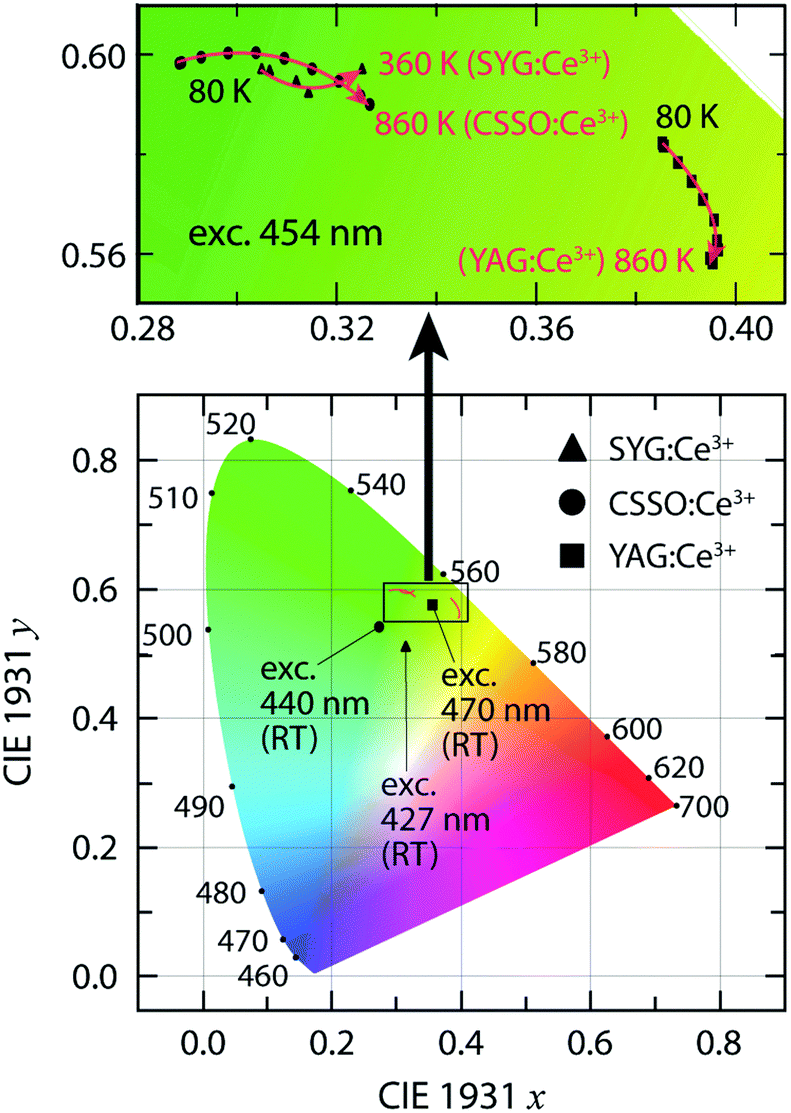 | ||
| Fig. 3 CIE 1931 color coordinate diagram for SYG:Ce3+, CSSO:Ce3+ and YAG:Ce3+, upon excitation at 427, 440, and 470 nm, respectively, at room temperature (RT). The variable temperature CIE coordinates of the three materials upon excitation at 454 nm are presented in detail in the upper image. | ||
The relation of the emission wavelengths between the three materials exhibits an opposite trend as one could expect based on the strength of their tetragonal crystal-field splitting (see ΔE5d1–2 in Table 1). A larger ΔE5d1–2 correlates with a more blue-shifted spectrum (smaller λ4f), which we interpret as the 2Eg barycenter of the lower 5d levels is ordered in energy as follows: CSSO:Ce3+ > SYG:Ce3+ > YAG:Ce3+, as indicated by the arrows in Fig. 2. The difference in the 2Eg barycenter may be related to the size-mismatch between Ce3+ (1.143 Å) and the substituting host atom, Ca2+ (1.12 Å), Y3+ (1.019 Å), and Sr2+ (1.27 Å), in 8-fold coordination.15 The size-mismatch ratios are approximately 2%, 10%, and 12% for CSSO:Ce3+, SYG:Ce3+, and YAG:Ce3+, respectively. Therefore, a larger size-mismatch appears to lower the energy level of the 2Eg barycenter, even though the local 8-fold environment surrounding the Ce3+ ions is less tetragonally distorted.
3.2 Temperature dependence of the emission spectra
To investigate the thermal stability of luminescence of the three garnet phosphors, both the emission intensity and the decay time of the Ce3+ emission were evaluated. It should be noted that because of the complexity of the various factors that can contribute to the temperature dependence of the luminescence intensity (e.g., temperature dependence of the absorption strength and temperature dependence of energy migration and reabsorption), an absolute, quantitative analysis of the (intrinsic) temperature quenching is difficult from the emission intensity data alone.22 Hence, for an evaluation of the intrinsic quenching process, luminescence decay time data are expected to provide a better, more accurate description.
Fig. 4(a–c) shows the emission spectra for excitation at 454 nm for all three samples over the temperature range of T = 80–860 K. Fig. 4(d) compares the temperature dependence of the emission intensity, as integrated over the energy region of 13000–22000 cm−1 and normalized to the maximum of the integrated intensity at T = 80–860 K. For CSSO:Ce3+, the integrated emission intensity exhibits a quite monotonic decrease upon increase of the temperature from T = 80 K up to T = 860 K. For SYG:Ce3+, the intensity increases from T = 80 K to approximately T = 240 K, which is followed by an abrupt decrease at T > 240 K until the luminescence intensity is virtually completely quenched at T ≈ 380–400 K. For YAG:Ce3+, the intensity shows an almost linear decrease between T = 80 K and T = 500 K, followed by a more pronounced decrease upon further temperature increase. Similar observations have been done in previous studies.22 The slow decrease of the luminescence intensity at T < 500 K is caused mainly by a reduction of the absorption strength of the 4f → 5d1 transition. This is because the second level of the 2F5/2 level is more thermally populated and the transition from this level to the lowest 5d level is symmetry forbidden.22,29
 | ||
| Fig. 4 Temperature dependence of the PL emission spectra and emission maxima [triangles, circles, and squares in (a–c)] of (a) SYG:Ce3+, (b) CSSO:Ce3+, and (c) YAG:Ce3+, upon excitation at 454 nm. (d) Temperature dependence of the normalized emission intensity for SYG:Ce3+, CSSO:Ce3+, and YAG:Ce3+. | ||
In contrast to the broadband emission spectra at RT (Fig. 2), the emission spectra at T = 80 K show a distinguished double peak signature [especially for CSSO:Ce3+, cf.Fig. 4(b)] corresponding to radiative 5d1 → 2F5/2 (higher energy) and 5d1 → 2F7/2 (lower energy) transitions, respectively. To determine the spectra of these two radiative transitions, two Gaussian functions were used to fit the doublet band in each spectrum, which reveals that the energy separation of the maxima of the 2F5/2 and 2F7/2 bands is approximately 1710 cm−1, 1410 cm−1, and 1560 cm−1 at T = 80 K for CSSO:Ce3+, SYG:Ce3+, and YAG:Ce3+, respectively. The energy difference between the 2F5/2 and 2F7/2 levels is comparable to the expected value of 2000 ± 250 cm−1 for Ce3+-doped garnet phosphors.11 The different values between CSSO:Ce3+, SYG:Ce3+, and YAG:Ce3+ is due to a difference in the spin–orbit coupling strength as Ce3+ ions interact with the neighboring atoms in the respective hosts.29 Furthermore, the emission maximum exhibits a shift toward longer wavelengths (red-shift) upon increasing temperature for all phosphors in the measured temperature range, see Fig. 4(a–c). Note that the emission maximum of SYG:Ce3+ is only shown up to T = 360 K since the spectra at T > 360 K are too weak to determine the emission maximum. The red-shift trend of the emission maximum upon temperature increase is in accordance with the temperature behavior of the color coordinates (Fig. 3). For YAG:Ce3+, the red-shift of the color coordinates persists up to around T = 600 K, which is in accordance with the results reported in ref. 19 and 30, whereas it turns to a blue-shift at T > 600 K. The blue-shift for YAG:Ce3+ at T > 600 K was not observed in previous studies due to limited highest temperatures (T ≤ 573 K).19,30 It is worth noting that the blue-shift of the color coordinates of YAG:Ce3+ from T = 600 K to T = 860 K (Fig. 3) is not evident for the emission maximum in the same temperature range [Fig. 4(c)]. The discrepancy between the color coordinates and the emission maximum originates from the way in which they have been obtained. For the calculation of the color coordinates x and y, the whole emission spectrum, which is determined by all spectral parameters such as the position, width and intensity of the 2F5/2 and 2F7/2 bands, is used. However, the emission maximum considers only the intensity of the superposition of the two bands. The latter implies that the emission maximum only contains partial information of the spectrum. This information depends on the extent of the spectral overlap between the 2F5/2 and 2F7/2 bands as explained by the following example: the emission maximum for CSSO:Ce3+ at T = 80 K [Fig. 4(b)] is mainly determined by the 2F5/2 band. Unlike the color coordinates, the emission maximum only reflects the emission color of part of the spectrum, particularly when the separation between the 2F5/2 and 2F7/2 bands is large. Therefore, to investigate the shift of the emission color as a function of temperature, the variation of the color coordinates, as shown in the upper image in Fig. 3, reflects the effect in a more accurate manner. It is also noted that the RT color coordinates (Fig. 3) are different between the ones derived from the emission spectra at RT in Fig. 2 and 4(a–c). This is likely an effect of that the measurements were performed with different excitation wavelengths.
On a fundamental level, the color shifting effect may be associated with a change in the tetragonal crystal field acting on the Ce3+ ions upon heating, e.g., through the increase of vibrationally induced tetragonal distortions of the local CeO8 moieties (red-shift), in competition with local thermal lattice expansion that, conversely, reduces the tetragonal crystal field (blue-shift).31 As the lattice size increases with elevating temperature, the local symmetry of CeO8 becomes more cubic-like due to the elongation of the Ce–O bonds.9 The red-shift may, however, be also explained by thermally activated energy migration and energy transfer to Ce3+ ions emitting at longer wavelength as the excitation and emission bands are thermally broadened.
3.3 Luminescence decay time measurements
Fig. 5(a–c) shows the luminescence decay curves for SYG:Ce3+, CSSO:Ce3+, and YAG:Ce3+ for excitation at 454 nm, at a selection of temperatures. Data for all temperatures are given in Fig. S1 (ESI†). All decay curves can be adequately approximated by single exponential fits. Fig. 5(d) compares the luminescence decay time, τ(T), over the measured temperature ranges, as extracted from the single exponential fits to the T dependent data in (a–c). In the low-temperature regime (T = 80–200 K), the luminescence decay time remains nearly constant, i.e. 52, 65, and 94 ns for SYG:Ce3+, CSSO:Ce3+, and YAG:Ce3+, respectively. The “intrinsic” decay time (lifetime) of the three Ce3+-doped garnet phosphors is within the range of the lifetime of Ce3+ luminescence (<100 ns) in many host materials.32 However, the lifetime of YAG:Ce3+ (94 ns) is higher than the values (60–67 ns) as reported in the literature by Bachmann et al.22 for YAG:Ce3+ for the Ce3+ dopant concentrations of 0.033–3.333%. Fig. 5(d) also compares the temperature dependent decay time of YAG:1%Ce3+ between this work and the one reported in ref. 22. The longer lifetime of YAG:Ce3+ observed here may be attributed to enhanced re-absorption processes due to a larger overlap between the excitation and emission spectra (Fig. 2), compared to the spectra reported by Bachmann et al.22 The difference in spectral overlap may stem from different synthesis details such as starting reagents and heat treatments, which may, for example, affect the distribution (homogeneity) of Ce3+ ions in the YAG host lattice, and lead to differences in the distance between Ce3+ ions. Such differences in the Ce–Ce distance should affect the energy transfer among Ce3+ ions, which shifts the position of the emission spectrum22 and hence alters the spectral overlap, i.e. the probability of re-absorption processes. | ||
| Fig. 5 (a) Temperature dependence of the luminescence decay curves for (a) SYG:Ce3+, (b) CSSO:Ce3+, and (c) YAG:Ce3+. (d) Luminescence decay time determined from the single-exponential fits in (a–c) as well as the decay time of YAG:1%Ce3+ from Bachmann et al.,22 as a function of temperature. Red solid curves are fits using a single-barrier quenching model.33,34 | ||
Upon further temperature increase (T > 200 K), the three materials show different behaviors. The onset temperature of thermal quenching for SYG:Ce3+, CSSO:Ce3+, and YAG:Ce3+ are at approximately T = 220 K, 600 K, and 400 K, respectively. In agreement with the recent study on CSSO:Ce3+,24 its luminescence decay time is approximately constant (71 ns) up to T = 600–700 K, which confirms an excellent internal thermal stability up to at least T = 600 K, although we observe a slightly different lifetime (65 ns) as compared to previous work.24 At T > 600 K, the decay time of CSSO:Ce3+ exhibits a relatively slow decrease from 64 ns at T = 600 K to 60 ns at T = 860 K, which further confirms that CSSO:Ce3+ has strong resistance against thermal quenching at high temperatures (up to at least T = 860 K). In contrast, the luminescence decay time of SYG:Ce3+ decreases abruptly from about 51 ns at T = 220 K to 29 ns at T = 300 K, and to 2 ns at T = 400 K at which the luminescence is nearly completely quenched. For YAG:Ce3+, the luminescence decay time decreases abruptly from τ = 86 ns at T = 400 K to τ = 63 ns at T = 600 K, and to τ = 9 ns at T = 860 K. The temperature at which the luminescence decay time has dropped to 80% of its “intrinsic” value, T80%, is approximately T = 280 K, T > 860 K, and T = 550 K, for SYG:Ce3+, CSSO:Ce3+, and YAG:Ce3+, respectively. The resistance to thermal quenching of luminescence for 5d1 → 4f emission for the three materials is hence ranked as follows, CSSO:Ce3+ > YAG:Ce3+ > SYG:Ce3+. It is worth noting that the T80% of YAG:Ce3+ with 1% doping concentration (T = 550 K) is almost 100 K lower than that (T = 645 K) of the same material synthesized with different starting reagents and heat treatments.22 This is in accordance with the longer lifetime obtained here (94 ns) compared to the one (≈61 ns) reported in ref. 22.
The overall thermal quenching behavior can be adequately fitted to a single-barrier quenching model [Fig. 5(d)], i.e. τ(T) = 1/[Γν + Γ0exp(−Ea/kBT)], where Γν is the radiative rate, Γ0 is the attempt rate for non-radiative processes, Ea is the activation energy for the thermal quenching processes, and kB is the Boltzmann constant.33,34 From a free fit, we obtain Ea of 0.33 ± 0.005 eV (SYG:Ce3+), and 0.45 ± 0.002 eV (YAG:Ce3+), see Table 1. In comparison, the Ea of thermal quenching of YAG:1%Ce3+ estimated from the data of Bachmann et al.22 is 0.53 ± 0.007 eV [Fig. 5(d)]. This is in good agreement with the one obtained here (0.45 eV), which suggests that the thermal quenching of luminescence of the two YAG:Ce3+ samples is driven by similar quenching process(es). As for CSSO:Ce3+, the decay time is almost constant up to T = 860 K (i.e., the highest applied T), which makes a free fit to the data unreliable. In order to estimate Ea for CSSO:Ce3+, we employed an empirical relation based on the results of SYG:Ce3+ and YAG:Ce3+, which shows that Ea is lowered by 26–62% with respect to E5d1-CB (Table 1), whose details are described in the next section. With this method, we estimate Ea to 0.5–1.0 eV for CSSO:Ce3+. In order to interpret in more detail these experimental findings, especially in regard to the positions of the ground and excited configurations of the Ce3+ dopants with respect to the valence and conduction bands of the respective hosts, we have constructed the VRBE diagrams of Ce3+ within SYG, CSSO, and YAG, as described in the following.
3.4 Vacuum referred binding energy diagrams
The way in which we have built the VRBE diagrams for the different hosts is based on published methods.11,35–43 The model, called the chemical shift model, provides electronic structure with absolute binding energies relative to the energies of the electron at rest, in vacuum.Using the spectroscopic data of the Ce3+ luminescence from the present work (Fig. 2 and Table 1) in comparison with previous studies, and the data for the Eu2+ and Eu3+ dopants in the studied hosts,11,44–46 the VRBE diagrams were constructed using input data for EV, Eex, EVC, ECT, U, λ5d1, and λ5d2, see Table 2. Fig. 6 compares the VRBE diagrams for the three hosts, as doped with Eu2+, Eu3+, or Ce3+. As seen in Table 2, the 4f-electron Coulomb repulsion energies U of Eu3+ in SYG (6.66 eV), CSSO (6.93 eV), and YAG (6.80 eV) are almost the same and are located in the 6.4–7.2 eV range, as typically observed for oxide materials.43 The major difference between the three phosphors lays in the other parameters, i.e. EV, Eex, EVC, ECT, λ5d1 (or D), and λ5d2 of the respective hosts, which affect the absolute location of the 5d levels with respect to the bottom of the CB (EC), see Fig. 6 and Table 2. The obtained energies (E5d1-CB) of the Ce3+ 5d1 level is 0.45 eV (SYG:Ce3+), 1.36 eV (CSSO:Ce3+), and 1.17 eV (YAG:Ce3+) below the CB, which are in good agreement with the results derived from the λ5d1 values, as reported previously [dashed lines in Fig. 6 and E5d1-CB (reference) in Table 2]. The larger E5d1-CB value means that the electron at the Ce3+ 5d1 level requires more thermal energy (higher temperature) to be promoted into the CB and the delocalized electron in the CB may be followed by charge trapping (luminescence is “killed”) at defect levels located within the band gap. This type of quenching process has been confirmed by measuring the photoconductivity of Y3Al2Ga3O12:Ce3+ and Y3Ga5O12:Ce3+ garnet phosphors when exciting the Ce3+ 4f electron to the 5d1 level as a function of temperature.47 In this context, a phosphor with a larger E5d1-CB is expected to exhibit higher stability against thermal quenching of luminescence, which is in good agreement with our results, cf. CSSO:Ce3+ (E5d1-CB = 1.36 eV, T80% > 860 K), YAG:Ce3+ (E5d1-CB = 1.17 eV, T80% = 550 K), and SYG:Ce3+ (E5d1-CB = 0.45 eV, T80% = 280 K), see Table 1. Similarly, for YAG:Ce3+, the separation between the Ce3+ 5d2 level and the bottom of the CB, as denoted by E5d2-CB, is much smaller than E5d1-CB. This suggests that, at a given temperature, more luminescence of Ce3+ ions with the excitation at λ5d2 is killed due to charge trapping at defects, as compared to the excitation at λ5d1. This is evident from a comparison of the intensity of the excitation bands centered at λ5d1 (stronger) and λ5d2 (weaker), see Fig. 2. The Ce3+ 5d2 levels of CSSO:Ce3+ and SYG:Ce3+ are located within the CB, which implies that the luminescence is completely quenched upon excitation at λ5d2. However, weak λ5d2 bands for CSSO:Ce3+ and SYG:Ce3+ can be still observed (Fig. 2). This may have its origin in a non-negligible amount of electrons in the CB returning to the 5d1 level, followed by 5d1 → 4f emission.
| VRBE parameters | SYG | CSSO | YAG |
|---|---|---|---|
| a Derived from the relation EVC = (1 + 8%)Eex.11 b Derived from the known ECe3+4f = −5.42 eV,44 and using the relation ECe3+4f = EEu2+4f − U + ΔE3+4f, where ΔE3+4f (=5.24 eV) is the difference between the 4f binding energies of Eu3+ and Ce3+.11 c Derived from the relation EC = EV + EVC,11 see arrow 1 in Fig. 6. d Derived from the relation EEu2+4f = EV + ECT,11 see arrow 2 in Fig. 6. e Derived from the relation EEu3+4f = EEu2+4f − U,11 see arrow 3 in Fig. 6. f Derived from the relation ECe3+4f = EEu3+4f + ΔE3+4f, where ΔE3+4f is defined in b,11 see arrow 4 in Fig. 6. g Derived from the relation ECe3+5di = ECe3+4f + 1240/λ5di, where i = 1 and 2,11 see arrow 5 in Fig. 6. h Derived from the relation D = ECe3+free − 1240/λ5d1, where ECe3+free = 6.12 eV.11 | |||
| • Input data, obtained from literature and Table 1 | |||
| E V (eV), top of valence band | −8.4344 | −8.9045 | −9.3811 |
| E ex (eV), exciton creation energy | 5.9044 a | 6.8145 a | 7.1011 |
| E VC (eV), band gap energy | 6.3744 | 7.3545 | 7.6711 a |
| E CT (eV), charge transfer energy of O2− → Eu3+ | 4.4344 | 4.8645 | 5.4211 |
| U (eV), 4f-electron Coulomb repulsion energy of Eu3+ | 6.6644b | 6.9345 | 6.8011 |
| λ 5d1 (nm), band maximum of 4f → 5d1 excitation (this work/reference) | 427/43544 | 440/45046 | 470/45711 |
| λ 5d2 (nm), band maximum of 4f → 5d2 excitation (this work/reference) | 297/30044 | 285/— | 340/34011 |
| • Output data, calculated using the input data above | |||
| E C (eV), bottom of conduction band c | −2.06 | −1.55 | −1.71 |
|
E
Eu2+4f (eV), 4f ground level of Eu2+d |
−4.00 | −4.04 | −3.96 |
|
E
Eu3+4f (eV), 4f ground level of Eu3+e |
−10.66 | −10.97 | −10.76 |
|
E
Ce3+4f (eV), 4f ground level of Ce3+f |
−5.42 | −5.73 | −5.52 |
| E Ce3+5d1 (eV), 5d1 excited level of Ce3+ (this work/reference)g | −2.51/−2.57 | −2.91/−2.97 | −2.88/−2.81 |
| E Ce3+5d2 (eV), 5d2 excited level of Ce3+ (this work/reference)g | −1.24/−1.29 | −1.38/— | −1.87/−1.87 |
| E 5d1-CB (eV), difference between ECe3+5d1 and EC (this work/reference) | 0.45/0.51 | 1.36/1.42 | 1.17/1.10 |
| E 5d2-CB (eV), difference between ECe3+5d2 and EC (this work/reference) | −0.82/−0.77 | −0.17/— | 0.16/0.16 |
| D (eV), red-shift of the lowest 5d level of Ce3+ (this work/reference)h | 3.22/3.27 | 3.30/3.36 | 3.48/3.41 |
 | ||
| Fig. 6 VRBE diagrams for the three hosts, left: SYG, middle: CSSO, and right: YAG, as doped with Eu2+, Eu3+, or Ce3+. The arrows 1, 2, 3, 4, and 5 represent the shifts of energy by amounts of EVC, ECT, U, ΔE3+4f, and energy of light with wavelength λ5d1 or λ5d2, respectively. Horizontal dashed lines (red color) represent the 5d1 and 5d2 levels of Ce3+ derived from the reported values of λ5d1 and λ5d2. Details of all the energy levels and VRBE parameters are listed in Table 2. | ||
4 Discussion
The three green-to-yellow-emitting phosphors SYG:Ce3+, CSSO:Ce3+, and YAG:Ce3+ have been shown to exhibit quite remarkably different luminescence properties, thus highlighting the important role of garnet host in regard to their optical performance. The nearly constant luminescence decay time (τ ≈ 60 ns) up to very high temperature, T = 860 K, for CSSO:Ce3+ is a unique feature as shown by the present host and is even better than the Na3–2xSc2(PO4)2:xEu2+ blue phosphor, which was recently claimed to be a zero-thermal-quenching phosphor up to T = 473 K.48 Moreover, the 4f–5d1 excitation and emission spectral characteristics of CSSO:Ce3+ make it a very good candidate for pc-WLEDs based on a blue LED, in particular, under a high-power operation which generates significant heat in pc-WLEDs, including the phosphor materials.The difference in intrinsic thermal stability of luminescence in SYG:Ce3+, CSSO:Ce3+, and YAG:Ce3+ may have its origin in different quenching processes in the three materials. On a fundamental level, the three primary processes that are thought to play a role in the thermal quenching of luminescence in Ce3+-doped materials exhibiting 5d–4f luminescence are (1) thermal ionization of the Ce3+ 5d electrons into the CB of the host crystal, followed by charge trapping at defects, (2) thermally activated nonradiative energy migration among Ce3+ ions to killer centers (generally known as concentration quenching), and (3) thermally activated crossover from the 5d excited state to the 4f ground state via electron–phonon coupling mechanisms.6,22,31,49 The fact that the Ce3+ concentration is identical (1%) in the three materials, but their stability against the thermal quenching of luminescence is remarkably different, suggests that concentration quenching is not responsible for the observed difference in the thermal quenching between the three phosphors. On the contrary, any of the other two processes are likely to play a significant role in determining the overall quenching behavior. The trend of increasing the thermal stability of luminescence (e.g. higher T80%) with increasing E5d1-CB values, see Table 1, would suggest that the quenching behavior is primarily determined by thermal ionization. However, the relatively large difference between E5d-CB and Ea shows that thermal ionization alone cannot determine the overall quenching behavior. Rather, these results point towards the contribution of both thermal ionization of the Ce3+ 5d electrons into the CB of the host crystal, followed by charge trapping, and thermally activated crossover from the 5d excited state to the 4f ground state via electron–phonon coupling mechanisms. In this context, we have previously shown that the excitation of the high-frequency phonon modes in the range of 600–900 cm−1 for YAG:Ce3+ promotes crossover non-radiative relaxation.31 For CSSO, this set of phonon modes are found at somewhat higher vibrational frequencies (800–900 cm−1),50 meaning that a higher temperature is needed for them to be thermally excited. Under the assumption that thermally activated 5d1 → 4f crossover contributes to the thermal quenching of luminescence in YAG:Ce3+ and CSSO:Ce3+, this would be in line with the observed difference in phonon frequencies. In comparison, the same set of phonon modes for SYG is located in the same frequency range as for YAG, i.e. 600–900 cm−1 (see Fig. S2, ESI†).51 This implies that the lower quenching temperature of SYG:Ce3+, see T80% in Table 1, is primarily attributed to its relatively smaller E5d1-CB (0.45 eV) as compared to that of YAG:Ce3+ (1.17 eV). In other words, this suggests that thermal quenching of luminescence in SYG:Ce3+ is driven primarily by thermal ionization rather than by 5d1 → 4f crossover relaxation.
The excellent thermal stability of CSSO:Ce3+ up to T = 860 K, suggests that it is a promising phosphor for application in efficient pc-WLEDs, which require a high thermal stability of luminescence up to at least T = 450 K (the temperature of the phosphors) in today's available LED packages.32Fig. 4(d) shows that the luminescence intensity of CSSO:Ce3+ at T = 450 K is about 26% lower with respect to that at T = 80 K. Since the decay time of CSSO:Ce3+ remains almost constant up to T = 860 K, this reduction of the luminescence intensity is mainly attributed to the decrease of the absorption strength of the 4f → 5d1 transition, as in the case for YAG:Ce3+.22 In comparison to YAG:Ce3+, which shows a decrease of the luminescence intensity by ≈37% at T = 450 K with respect to that at T = 80 K [Fig. 4(d)], CSSO:Ce3+ shows a better thermal stability in terms of luminescence intensity, in agreement with the literature.52 The difference in thermal stability of luminescence intensity between CSSO:Ce3+ and YAG:Ce3+ tends to become larger at higher temperatures, e.g. differences of 11% at T = 450 K, and 28% at T = 600 K [Fig. 4(d)], which suggests that CSSO:Ce3+ is a better candidate for high power pc-WLEDs.
Fig. 7(a) shows the PL emission spectra of the pc-WLED prototype. The prototype consists of the CSSO:Ce3+ phosphor deposited on a commercial InGaN blue LED (λ ≈ 420 nm) under a voltage of 3.4, 3.6, 3.8, and 4.0 V, respectively. The integrated intensity of the emitted light of the pc-WLED prototype shows a linear increase with increasing applied voltage [Fig. 7(b)]. This suggests that the variation of the temperature of the blue LED chip upon applied voltage within the typical operating range for InGaN LEDs (3.4–4.0 V) has no significant impact on the strength of the 4f–5d transition (both blue absorption and green emission) of CSSO:Ce3+ when coated on top of the LED chip. This agrees with the observed excellent thermal stability of the emission intensity of CSSO:Ce3+ [Fig. 4(d)]. Under an applied voltage of 3.4 V, the prototype exhibits a luminous efficacy of optical radiation (LER) of 243 lm W−1 (≈4.191 × 10−3 lm/1.725 × 10−5 W, defined as the ratio of the total luminous flux to radiant power); the inset in Fig. 7(a) shows a photograph of the pc-WLED prototype under this bias. The prototype has a color rendering index (CRI) of 58, which is calculated using the Python colour science package called “Colour” with the measured spectral power distribution [Fig. 7(a)].53 In comparison with other pc-WLEDs with similar CRIs (57–63), the LER obtained here is generally lower than the others (370–430 lm W−1),54 which may be caused by a larger fraction of blue light leaking through the phosphor.28 This relatively strong blue light is also evident from the CIE coordinates of the pc-WLED prototype that lay near the region of bluish white color or blue color (Fig. 8). Thus, the light emitted from the prototype is perceived as cold white light. The prototype exhibits a CCT = 13604 K, which may be compared to that of the well known system comprising a blue LED and YAG:Ce3+ (CCT = 5600 K),28 see Fig. 8. The white balance and the CRI of the pc-WLED prototype may thus be improved by the addition of a red-emitting phosphor and/or red-emitting core/shell quantum dots to the present system, as implemented in other studies.28,52,55
 | ||
| Fig. 7 (a) Emission spectra of the pc-WLED prototype coated with CSSO:Ce3+ under operation with a voltage of 3.4 V, 3.6 V, 3.8 V, and 4.0 V, respectively. Inset: Bluish white emission from the pc-WLED operated with 3.4 V. (b) Integrated intensity of the emission spectrum of the pc-WLED prototype as a function of applied voltage. | ||
 | ||
| Fig. 8 CIE 1931 color coordinate diagram for the pc-WLED prototype based on CSSO:Ce3+ and a blue LED [CIE 1931 xy = (0.220, 0.324), and CCT = 13604 K] as derived from the spectrum of the prototype operated with 3.4 V in Fig. 7(a), which is compared to the one based on YAG:Ce3+ and a blue LED [CIE 1931 xy = (0.330,0.339), and CCT = 5600 K]. | ||
5 Conclusions
To conclude, we have investigated the luminescence properties of the three garnet type phosphors CSSO:Ce3+, SYG:Ce3+ and YAG:Ce3+. By monitoring the temperature dependence of the luminescence decay time, we establish an excellent, intrinsic, thermal stability of luminescence in CSSO:Ce3+ with a nearly constant decay time (≈60 ns) up to T = 860 K. In comparison, SYG:Ce3+ and YAG:Ce3+ exhibit a significant reduction of the luminescence decay time, starting at around T = 280 K and T = 550 K, respectively, suggesting a lower internal thermal stability of luminescence in these materials. These experimental results are supported by the energy separation between the Ce3+ 5d1 level and the CB of the respective hosts, which are found at 1.36 eV (CSSO:Ce3+), 0.45 eV (SYG:Ce3+), and 1.17 eV (YAG:Ce3+), respectively, as predicted by their VRBE diagrams. The performance of CSSO:Ce3+ was further evaluated by applying the phosphor on a blue InGaN LED. The system shows a luminous efficacy of optical radiation of 243 lm W−1 and a linear response with increasing applied voltage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Swedish Research Council Formas (Grant No. 2013-1723), and the European Commission for funding through the Marie Curie Initial Training network LUMINET (Grant No. 316906) is gratefully acknowledged. We also thank Erica Viviani, University of Verona, for expert technical assistance, and Jumpei Ueda, Kyoto University, for fruitful discussions.References
- S. Pimputkar, J. S. Speck, S. P. DenBaars and S. Nakamura, Nat. Photonics, 2009, 3, 180 CrossRef.
- T. Suehiro, N. Hirosaki and R.-J. Xie, ACS Appl. Mater. Interfaces, 2011, 3, 811–816 CrossRef PubMed.
- T.-C. Liu, B.-M. Cheng, S.-F. Hu and R.-S. Liu, Chem. Mater., 2011, 23, 3698–3705 CrossRef.
- S. Nakamura, MRS Bull., 2009, 34, 101–107 CrossRef.
- S. Ye, F. Xiao, Y. Pan, Y. Ma and Q. Zhang, Mater. Sci. Eng., R, 2010, 71, 1–34 CrossRef.
- Y.-C. Lin, M. Karlsson and M. Bettinelli, Top. Curr. Chem., 2016, 374(21), 1–47 Search PubMed.
- Z. Xia and A. Meijerink, Chem. Soc. Rev., 2017, 46, 275–299 RSC.
- Z. Xia and Q. Liu, Prog. Mater. Sci., 2016, 84, 59–117 CrossRef.
- J. L. Wu, G. Gundiah and A. Cheetham, Chem. Phys. Lett., 2007, 441, 250–254 CrossRef.
- A. Kalaji, P. J. Saines, N. C. George and A. K. Cheetham, Chem. Phys. Lett., 2013, 586, 91–96 CrossRef.
- P. Dorenbos, J. Lumin., 2013, 134, 310–318 CrossRef.
- P. Dorenbos, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 235110 CrossRef.
- N. C. George, K. A. Denault and R. Seshadri, Annu. Rev. Mater. Res., 2013, 43, 481–501 CrossRef.
- N. C. George, A. J. Pell, G. Dantelle, K. Page, A. Llobet, M. Balasubramanian, G. Pintacuda, B. F. Chmelka and R. Seshadri, Chem. Mater., 2013, 25, 3979–3995 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- P. Ghigna, S. Pin, C. Ronda, A. Speghini, F. Piccinelli and M. Bettinelli, Opt. Mater., 2011, 34, 19–22 CrossRef.
- F. Pan, M. Zhou, J. Zhang, X. Zhang, J. Wang, L. Huang, X. Kuang and M. Wu, J. Mater. Chem. C, 2016, 4, 5671–5678 RSC.
- S. J. Marin, M. O'Keeffe, V. G. Young and R. B. V. Dreele, J. Solid State Chem., 1991, 91, 173–175 CrossRef.
- C. C. Lin and R.-S. Liu, J. Phys. Chem. Lett., 2011, 2, 1268–1277 CrossRef PubMed.
- H. Daicho, T. Iwasaki, K. Enomoto, Y. Sasaki, Y. Maeno, Y. Shinomiya, S. Aoyagi, E. Nishibori, M. Sakata and H. Sawa, et al. , Nat. Commun., 2012, 3, 1132 CrossRef PubMed.
- H. Zhu, C. C. Lin, W. Luo, S. Shu, Z. Liu, Y. Liu, J. Kong, E. Ma, Y. Cao and R.-S. Liu, et al. , Nat. Commun., 2014, 5, 4312 CrossRef PubMed.
- V. Bachmann, C. Ronda and A. Meijerink, Chem. Mater., 2009, 21, 2077–2084 CrossRef.
- P. F. Smet, A. B. Parmentier and D. Poelman, J. Electrochem. Soc., 2011, 158, R37–R54 CrossRef.
- I. Berezovskaya, Z. Khapko, A. Voloshinovskii, N. Efryushina, S. Smola and V. Dotsenko, J. Lumin., 2018, 195, 24–30 CrossRef.
- W. B. Im, N. George, J. Kurzman, S. Brinkley, A. Mikhailovsky, J. Hu, B. F. Chmelka, S. P. DenBaars and R. Seshadri, Adv. Mater., 2011, 23, 2300–2305 CrossRef PubMed.
- A. Kaminska, A. Duzynska, M. Berkowski, S. Trushkin and A. Suchocki, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155111 CrossRef.
- A. A. Setlur, W. J. Heward, Y. Gao, A. M. Srivastava, R. G. Chandran and M. V. Shankar, Chem. Mater., 2006, 18, 3314–3322 CrossRef.
- M. R. Krames, O. B. Shchekin, R. Mueller-Mach, G. O. Mueller, L. Zhou, G. Harbers and M. G. Craford, J. Disp. Technol., 2007, 3, 160–175 Search PubMed.
- D. Robbins, J. Electrochem. Soc., 1979, 126, 1550–1555 CrossRef.
- C.-C. Chiang, M.-S. Tsai and M.-H. Hon, J. Electrochem. Soc., 2008, 155, B517–B520 CrossRef.
- Y.-C. Lin, P. Erhart, M. Bettinelli, N. C. George, S. F. Parker and M. Karlsson, Chem. Mater., 2018, 30, 1865–1877 CrossRef.
- A. Setlur, Electrochem. Soc. Interface, 2009, 16, 32 Search PubMed.
- S. Shionoya, W. M. Yen and H. Yamamoto, Phosphor handbook, CRC press, 2006 Search PubMed.
- B. Di Bartolo, Advances in Nonradiative Processes in Solids, Springer My Copy, UK, 2014 Search PubMed.
- P. Dorenbos, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 165107 CrossRef.
- P. Dorenbos, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 035118 CrossRef.
- P. Dorenbos, J. Electrochem. Soc., 2005, 152, H107–H110 CrossRef.
- P. Dorenbos and E. G. Rogers, ECS J. Solid State Sci. Technol., 2014, 3, R150–R158 CrossRef.
- E. Rogers and P. Dorenbos, J. Lumin., 2014, 153, 40–45 CrossRef.
- E. Rogers and P. Dorenbos, ECS J. Solid State Sci. Technol., 2014, 3, R173–R184 CrossRef.
- E. Rogers and P. Dorenbos, J. Lumin., 2014, 155, 135–140 CrossRef.
- P. Dorenbos, J. Lumin., 2013, 135, 93–104 CrossRef.
- P. Dorenbos, ECS J. Solid State Sci. Technol., 2013, 2, R3001–R3011 CrossRef.
- H. Luo, L. Ning, Y. Dong, A. J. Bos and P. Dorenbos, J. Phys. Chem. C, 2016, 120, 28743–28752 CrossRef.
- L. Zhou, W. Zhou, F. Pan, R. Shi, L. Huang, H. Liang, P. A. Tanner, X. Du, Y. Huang and Y. Tao, et al. , Chem. Mater., 2016, 28, 2834–2843 CrossRef.
- Y. Liu, X. Zhang, Z. Hao, Y. Luo, X. Wang and J. Zhang, J. Mater. Chem., 2011, 21, 16379–16384 RSC.
- J. Ueda, S. Tanabe and T. Nakanishi, J. Appl. Phys., 2011, 110, 053102 CrossRef PubMed.
- Y. H. Kim, P. Arunkumar, B. Y. Kim, S. Unithrattil, E. Kim, S.-H. Moon, J. Y. Hyun, K. H. Kim, D. Lee and J.-S. Lee, et al. , Nat. Mater., 2017, 16, 543 CrossRef PubMed.
- J. Ueda, P. Dorenbos, A. J. Bos, A. Meijerink and S. Tanabe, J. Phys. Chem. C, 2015, 119, 25003–25008 CrossRef.
- F. Piccinelli, A. Speghini, G. Mariotto, L. Bovo and M. Bettinelli, J. Rare Earths, 2009, 27, 555–559 CrossRef.
- Y.-C. Lin, P. Erhart, M. Bettinelli and M. Karlsson, to be published.
- Y. Shimomura, T. Honma, M. Shigeiwa, T. Akai, K. Okamoto and N. Kijima, J. Electrochem. Soc., 2007, 154, J35–J38 CrossRef.
- Color rendering index was calculated using the Python colour science package called Colour developed by JetBrains PyCharm, http://colour-science.org/.
- C.-C. Sun, Y.-Y. Chang, T.-H. Yang, T.-Y. Chung, C.-C. Chen, T.-X. Lee, D.-R. Li, C.-Y. Lu, Z.-Y. Ting and B. Glorieux, et al. , J. Solid State Light., 2014, 1, 19 CrossRef.
- S. Abe, J. J. Joos, L. I. Martin, Z. Hens and P. F. Smet, Light: Sci. Appl., 2017, 6, e16271 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tc02907e |
| ‡ Current address: Institute of Applied Physics, TU Bergakademie Freiberg, Leipziger Str. 23, 09596 Freiberg, Germany. |
| § Current address: Advanced Technology Institute, Department of Electrical and Electronic Engineering, University of Surrey, Guildford GU2 7XH, UK. |
| This journal is © The Royal Society of Chemistry 2018 |