
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly stable doping of a polar polythiophene through co-processing with sulfonic acids and bistriflimide†
Anna I.
Hofmann
 *,
Renee
Kroon
,
Liyang
Yu
and
Christian
Müller
*
*,
Renee
Kroon
,
Liyang
Yu
and
Christian
Müller
*
Department of Chemistry and Chemical Engineering, Chalmers University of Technology, 41296 Göteborg, Sweden. E-mail: hofanna@chalmers.se; christian.muller@chalmers.se
First published on 22nd June 2018
Abstract
Doping of organic semiconductors is currently an intensely studied field, since it is a powerful tool to optimize the performance of various organic electronic devices, ranging from organic solar cells, to thermoelectric modules, and bio-medical sensors. Despite recent advances, there is still a need for the development of highly conducting polymer:dopant systems with excellent long term stability and a high resistance to elevated temperatures. In this work we study the doping of the polar polythiophene derivative p(g42T-T) by various sulfonic acids and bistriflimide via different processing techniques. We demonstrate that simple co-processing of p(g42T-T) with an acid dopant yields conductivities of up to 120 S cm−1, which remain stable for more than six months under ambient conditions. Notably, a high conductivity is only achieved if the doping is carried out in air, which can be explained with a doping process that involves an acid mediated oxidation of the polymer through O2. P(g42T-T) doped with the non-toxic and inexpensive 1,3-propanedisulfonic acid was found to retain its electrical conductivity for at least 20 hours upon annealing at 120 °C, which allowed the bulk processing of the doped polymer into conducting, free-standing and flexible films and renders the di-acid a promising alternative to commonly used redox dopants.
Organic semiconductors attract a great deal of attention as a class of materials that promises to pave the way towards the cost efficient large-scale production of light, flexible and bio-compatible electronic devices and opens up a world of possibilities for innovative technologies, which cannot be realized using “traditional”, inorganic semiconductors. Of particular interest for such devices are semiconducting polymers, not only owing to their excellent mechanical properties but also due to the ease of processing from solution or melt. The electronic properties of semiconducting polymers depend strongly on the charge carrier density, which can be tuned by so-called doping. Hence, doping of semiconducting polymers is currently a topic of intense research,1 needed to optimize various electronic devices, ranging from field-effect transistors2,3 to solar cells4–6 and thermoelectric generators.7,8
Doping of semiconducting polymers can be achieved either via electron transfer between a dopant molecule and the polymer or via a proton/hydride transfer from the dopant to the polymer backbone. In case of the polythiophene derivatives poly(3-hexylthiophene) (P3HT) and poly(2,5-bis(3-tetradecylthiophene-2-yl)thieno[3,2-b]thiophene) (PBTTT), for instance, the conductivity can be enhanced by several orders of magnitude upon p-doping with the oxidizing agent 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) or with strong acids and can reach up to 103 S cm−1.9–15 However, such high conductivities are only obtained by sequential doping of thin P3HT and PBTTT films, since the mixing of polymer and dopant in solution leads to aggregation of the doped polymers, which affects the nanostructure of the polymer films and commonly entails a significant decrease of the film conductivity to typically not more than 1 S cm−1.12,16–18 For a more facile and cost-efficient fabrication of doped polythiophene structures, it is desirable to develop strategies that make it possible to dope the polymers using simple one-pot processes or bulk processing methods, without compromising on the conductivity of the polymers.
One possible approach to improve the processability of doped polythiophenes is the design of self-doped systems via the introduction of ionic side chains to the polymer backbone. Such self-doped polythiophene derivatives are water soluble and can display conductivities of approximately 10 S cm−1.19,20 More recent studies have demonstrated that the introduction of polar side chains enhances significantly the processability and miscibility of several p-type and n-type organic semiconductor/dopant systems.1,21–26 The polar polythiophene derivative p(g42T-T), for instance, (see Fig. 1 for chemical structure) shows enhanced solubility in organic solvents, which makes it possible to co-process p(g42T-T) with the dopant F4TCNQ to obtain homogeneous films with a conductivity of up to 100 S cm−1 and an improved thermal stability as compared to P3HT:F4TCNQ.21 Nevertheless, it can be anticipated that the high vapour pressure of neutral F4TCNQ and the reversible nature of the doping reaction eventually lead to the sublimation of F4TCNQ,1 which will compromise the long-term stability of the doped system. Furthermore, it has to be considered that the toxicity and the considerable price of F4TCNQ are limiting factors for many of the potential applications. Therefore, alternative doping methods with improved thermal stability, using inexpensive and non-toxic dopants are highly sought after.
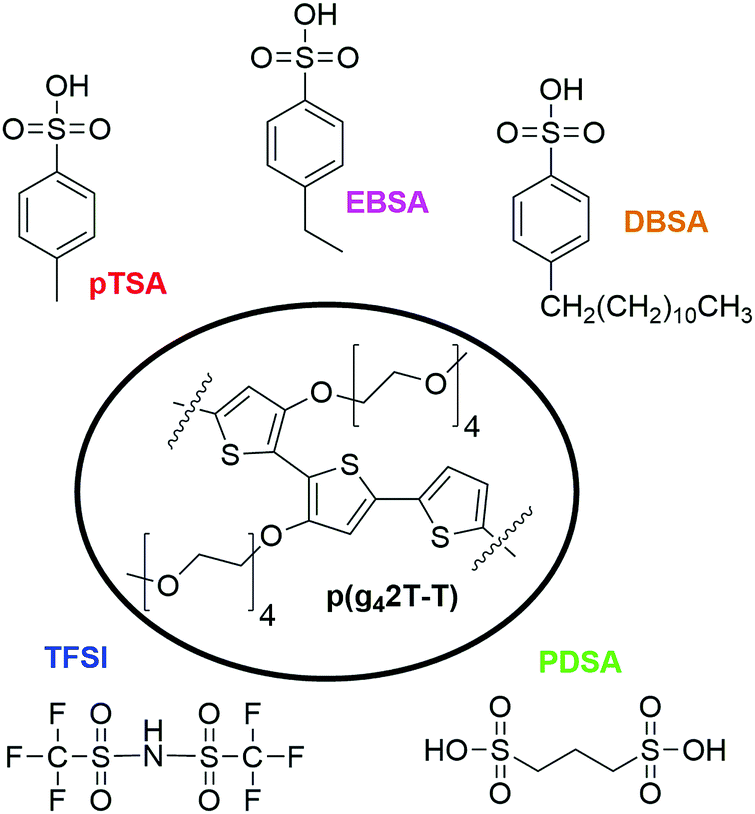 | ||
| Fig. 1 Chemical structure of the p(g42T-T) polymer and the acid dopants p-toluenesulfonic acid (pTSA), 4-ethylbenzenesulfonic acid (EBSA), 4-dodecylbenzenesulfonic acid (DBSA), trifluoromethanesulfonimide (TFSI) and 1,3-propanedisulfonic acid (PDSA). | ||
In this paper, we demonstrate that p(g42T-T) can be efficiently doped by various sulfonic acids and by bistriflimide. By comparing sequentially doped and co-processed p(g42T-T) we found that co-processing results in films with a superior electrical conductivity of up to 120 S cm−1 that remained stable for at least six months at ambient conditions. Upon doping with the di-acid 1,3-propanedisulfonic acid p(g42T-T) displayed an excellent thermal stability towards annealing at 120 °C for 20 hours, which allowed the solid state pressing of the doped polymer into a free-standing, conducting film.
The chemical structures of the p(g42T-T) polymer and the five different acid dopants used for this work are displayed in Fig. 1. Since the ability of acids to dope conjugated polymers increases with their acid strength,27 we chose to work with five acids which are known, or can be expected to be strong acids with pKa values <0. By comparing p(g42T-T) films doped with p-toluenesulfonic acid (pTSA) (pKa −1.3), 4-ethylbenzenesulfonic acid (EBSA) and 4-dodecylbenzenesulfonic acid (DBSA) the effect of the molecular size of the dopant was studied. Doping with the di-acid 1,3-propanedisulfonic acid (PDSA) allowed to investigate the impact of the number of sulfonic acid groups per dopant molecule, whereas the comparison with bis(trifluoromethane)sulfonimide (TFSI) (pKa −1228) doped p(g42T-T) gave information on the importance of the acid functionality.
In order to determine the most promising processing route, p(g42T-T) was doped with pTSA and TFSI via exposure of the polymer film to acid vapour, immersion into a solution of the respective acid in acetonitrile or via co-processing of the polymer and the acid from a mutual solvent. All three doping methods lead to an increase of the conductivity by several orders of magnitude as compared to the conductivity of the pristine p(g42T-T). We find, that the resulting conductivities were very similar for both acid dopants but depended strongly on the processing route (Fig. 2a). For sequentially solution doped p(g42T-T), conductivities of up to 10 S cm−1 were obtained upon optimisation of the dipping time (Fig. S1, ESI†), whereas sequential vapour doping resulted in much higher conductivities in the range of 70 to 100 S cm−1. It is noteworthy that simple co-processing of p(g42T-T) with the acid dopants from a mutual solvent yielded a conductivity of up to 120 S cm−1.
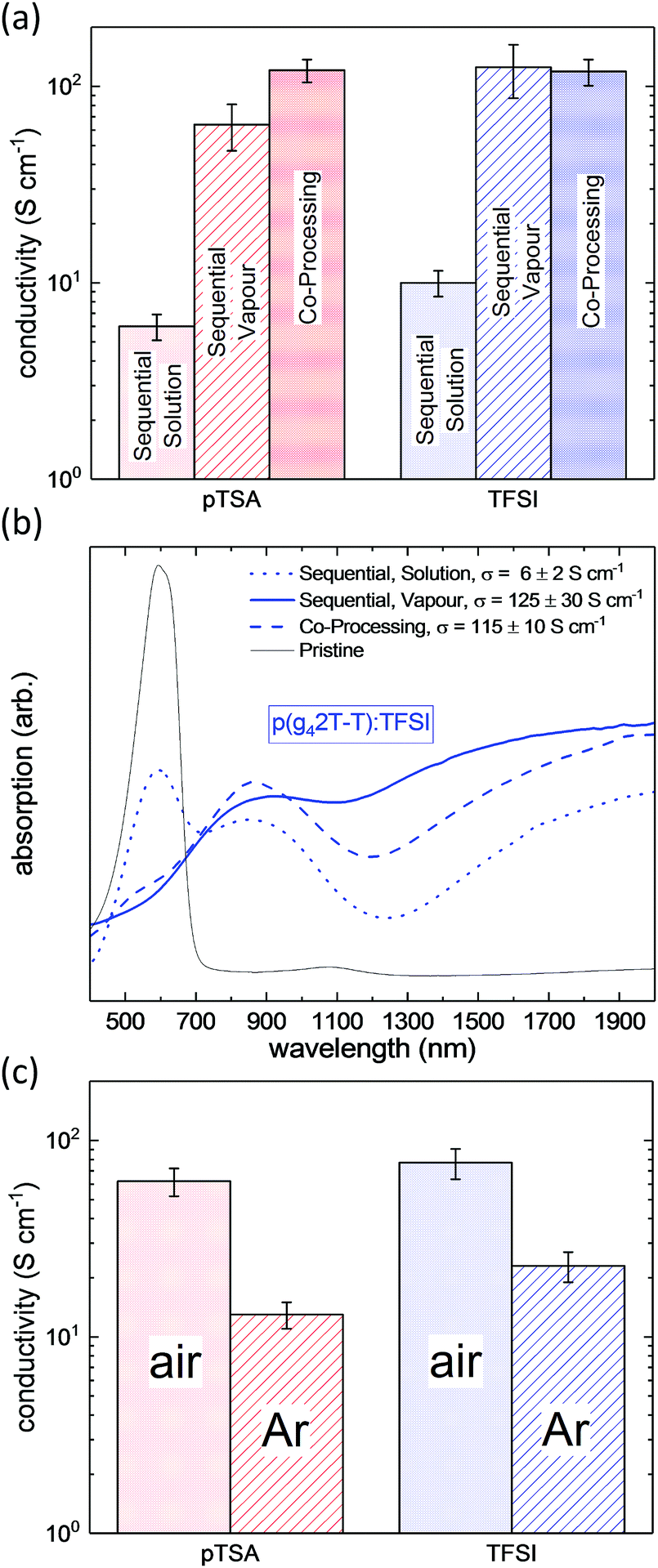 | ||
| Fig. 2 (a) Electrical conductivity and (b) absorption spectra of p(g42T-T) films, doped with pTSA and TFSI via sequential doping in acid solution or vapour, or via co-processing, obtained using optimised doping processes and (c) conductivity of p(g42T-T):TFSI and p(g42T-T):pTSA, co-processed from degassed solution and dried in air or under argon atmosphere, respectively. | ||
To probe the doping of the p(g42T-T) films, UV-Vis-NIR spectroscopy was performed. As exemplarily shown for p(g42T-T):TFSI in Fig. 2b, all of the doped polymer films displayed pronounced sub-band-gap absorption at 900 nm and in the IR regime, which is characteristic for polaronic absorption and confirms the doping of p(g42T-T). However, the sequentially solution doped film exhibited prominent absorption at 600 nm, which corresponds to the absorption maximum of the neat polymer and indicates a lower degree of doping. Thus, the lower conductivity of sequentially solution doped p(g42T-T) can be directly related to its lower degree of doping. Therefore, we can state that sequential doping from solution was the least efficient processing route, whereas vapour doping and co-processing resulted in comparable or equally high conductivities for pTSA and TFSI doped p(g42T-T), respectively. Thus, our method of choice for further studies on acid doping of p(g42T-T) was co-processing, as it combined high electrical conductivity with the ease of processing and a precise control over the amount of dopant molecules that is introduced to the system.
To elucidate the role of oxygen in the doping process we compared the conductivity of pTSA and TFSI doped p(g42T-T) films, which had been fabricated either under ambient conditions or in the absence of oxygen (see Fig. 2c). We found, that processing in air systematically yielded a 3 to 5 times higher conductivity, which indicates that the acid doping process involves an acid mediated oxidation of the polymer through O2.27,29 Based on these findings all following experiments were performed in air.
In order to select the most suitable acid dopant, p(g42T-T) was co-processed in solution with different concentrations of either pTSA, EBSA, DBSA, PDSA or TFSI and then drop casted into films.
One of the main challenges for the co-processing of conjugated polymers is, that the doped polymers tend to strongly aggregate in solution. However, upon addition of the acids to the p(g42T-T) solution no precipitation was observed, with the exception of p(g42T-T) solutions containing more than 20 mol% TFSI. A gelation of the polymer solution, which we attribute to the reduced solubility of the polymer upon doping, was observed only about one hour after the admixing of the acid dopant and provided a convenient time window for processing. Any concerns that the acidic conditions could result in the cleavage of the tetraethylene glycol side chains of p(g42T-T) were shown to be unfounded since the solubility of p(g42T-T) was fully recovered after de-doping of the polymer in alkaline conditions.
In order to follow the doping of the different p(g42T-T):acid systems, UV-Vis-NIR spectroscopy was performed. Fig. 3a displays the absorption spectra of TFSI and pTSA doped p(g42T-T), recorded as a function of the dopant concentration. It can be seen that the absorption of the pristine polymer at 600 nm diminished with increasing dopant concentration, whereas the polaronic absorption in the near IR region rose. The same characteristic features were observed in the absorption spectra of p(g42T-T) in the presence of EBSA, DBSA and PDSA, which confirmed that all five acids were able to dope p(g42T-T) (see Fig. S2 and S3, ESI†).
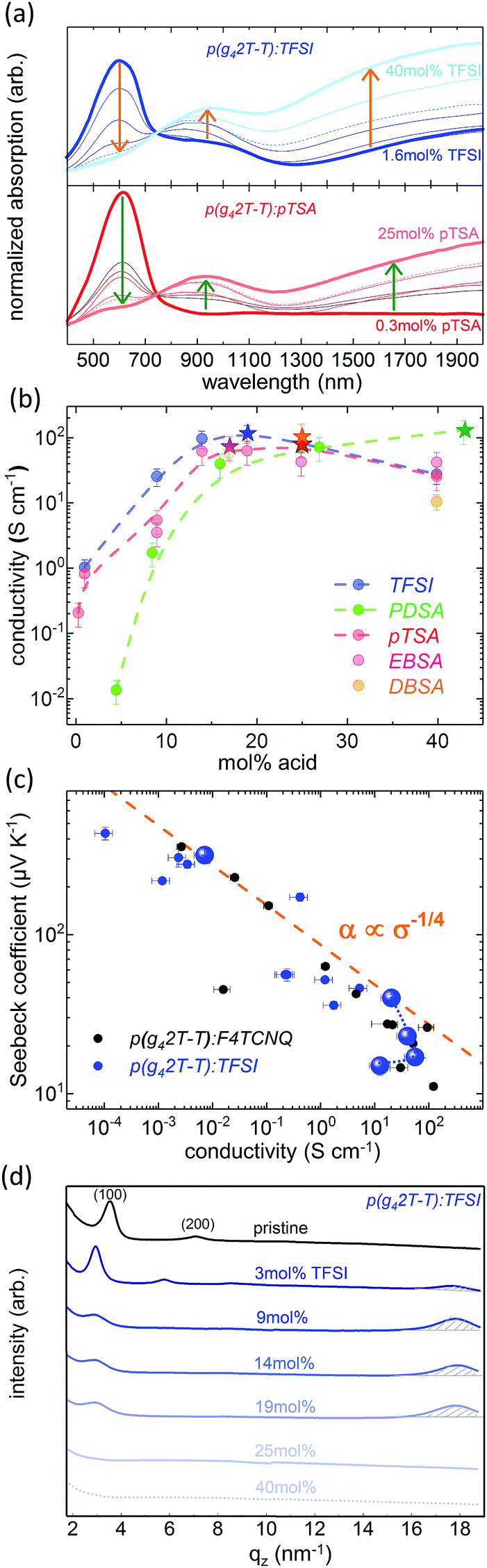 | ||
| Fig. 3 (a) UV-Vis-NIR absorption spectra of p(g42T-T):TFSI and p(g42T-T):pTSA as a function of dopant concentration, normalised to the absorption at 745 nm. (b) Average conductivity of drop cast p(g42T-T) co-processed with different acids as a function of the dopant concentration (dashed lines are a guide to the eye); stars denote composition at which the maximum conductivity is reached for each dopant. (c) Seebeck coefficient as a function of conductivity of p(g42T-T):TFSI, the dashed line represents the empirical correlation α ∝ σ−1/4 proposed by Glaudell et al.,16 highlighted data points serve as guide to the eye illustrating the evolution of α and σ at high dopant concentrations. (d) Line cuts along the out of plane axis qz of GIWAXS spectra as a function of dopant concentration for p(g42T-T):TFSI films. | ||
Monitoring of the UV-Vis-NIR spectrum of a p(g42T-T):PDSA solution over time revealed that the doping level continued to increase for several days (see Fig. S3, ESI†). Furthermore, we observed that the conductivity of all acid doped films cast at room temperature attained its maximum only one day after the film preparation (see Fig. S3, ESI†), indicating that the kinetics of the doping reaction are slow. This corroborates, that the doping mechanism is not (solely) based on an acid–base reaction between the dopant and the polymer backbone, but involves an oxidation reaction of the polymer with the ambient O2. However, it was found that the doping process could be accelerated by a short annealing of the film.
The maximum film conductivity was found to be comparable for all co-processed p(g42T-T):acid systems. At low dopant concentrations, the electrical conductivity increased several orders of magnitude with increasing dopant concentration until a conductivity maximum in the range of 110 S cm−1 ± 10 S cm−1 was reached. For all mono-acidic dopants, the conductivity maximum was attained at dopant concentrations between 15 and 25 mol%, whereas upon doping with the di-acid PDSA the maximum conductivity was only reached at more than 40 mol% of dopant (see Fig. 3b). This indicates that a considerable amount of the PDSA functionalities stay unreacted and that dopant molecules are present in excess.
With further increasing dopant concentration, the conductivity of the p(g42T-T):acid systems diminished. However, the UV-Vis-NIR absorption spectra did not reveal a decrease in doping for these samples. Thus, the drop in conductivity most likely results from the high percentage of insulating matter in the material, namely the dopant, which disrupts the nanostructure of the conjugated polymer and affects the charge transport through the film.
This hypothesis was confirmed by the analysis of the Seebeck coefficient α of acid doped p(g42T-T) as a function of its conductivity σ (see Fig. 3c). At low and intermediate dopant concentrations the Seebeck coefficient decreased with increasing doping, following a α ∝ σ−1/4 trend, as proposed by Glaudell et al.16 At high dopant concentrations, however, a further increase of the dopant concentration resulted in a decrease of the electrical conductivity, while the Seebeck coefficient was not significantly affected, leading to a strong deviation from the α ∝ σ−1/4 correlation. This observation indicates that the system is mobility limited by nonconducting domains of excess dopant.30
This was furthermore corroborated by grazing-incidence wide-angle X-ray scattering (GIWAXS) experiments, which revealed a change of the nanostructure of thin p(g42T-T) films as a function of the dopant concentration (see Fig. 3d and Fig. S4, ESI†). As reported previously,21 pristine p(g42T-T) films contain ordered domains of edge-on oriented lamellae (h00) with a packing distance of 1.8 nm ((100) reflection at qz = 3.5 nm−1). With increasing concentration of the acid dopant, the lamellar structure evolved into a mixed edge- and face-on configuration and the packing distance increased to 2.2 nm, which can be explained by the incorporation of the acid molecules in the lamellar structure of the polymer crystals. Furthermore, the increase in doping concurred with the rise of a strong scattering peak at qz = 17.8 nm−1 (d = 0.4 nm), indicating a high degree of π–π stacking between the doped and therefore planarized polythiophene moieties of the highly conducting p(g42T-T) (see Fig. 3d and Fig. S4, ESI†). However, at very high dopant concentration both scattering peaks decreased drastically, indicating that the lamellar structure as well as the π–π stacking was disrupted. A lower degree of order commonly obstructs the charge transport within a material and thus explains a decrease of its electrical conductivity.30–33
The comparison of the GIWAXS spectra of p(g42T-T) doped with different acids indicates that doping with the super-acid TFSI has the highest influence on the nanostructure, since the most intense π–π-stacking peak in p(g42T-T):TFSI is reached at a lower dopant concentration (19 mol%) as compared to p(g42T-T):pTSA (25 mol%), p(g42T-T):EBSA (25 mol%) and p(g42T-T):DBSA (40 mol%). Nevertheless, for the different acid dopants no strong influence on the macroscopic electrical conductivity of the doped p(g42T-T) films was observed. This suggests that for the doping of polythiophene derivatives one may choose from a wide palette of strong acids, which yield comparable conductivities of the polymer films. Thus, acid doping is a very promising strategy considering the diversity of possible dopant molecules, which allows to take into account not only factors such as availability and cost, but also specific processing parameters and stability requirements.
The thermal stability of the electronic properties of doped polymers, for instance, is a crucial parameter for many processing methods and applications. Therefore, we monitored the conductivity of acid doped p(g42T-T) upon annealing of the doped films in air at various temperatures. All acid doped p(g42T-T) films exhibited excellent resistance to short term exposure of less than 5 min to temperatures up to 180 °C and retained their electronic properties during annealing at 120 °C for 10 hours or longer (see Fig. 4 and Fig. S5, ESI†). Upon exposure to temperatures higher than 120 °C, pTSA, EBSA and DBSA doped p(g42T-T) films displayed similar degradation of their conductivity, which dropped to half of the initial value after 45 min annealing at 140 °C (see Fig. S5, ESI†). P(g42T-T) doped with TFSI, however, showed a better temperature stability and retained half the initial conductivity for 1.5 hours of annealing at 140 °C (see Fig. 4b). The highest temperature stability was obtained for PDSA doped p(g42T-T), which preserves 50% of its initial conductivity for more than 3 hours upon annealing at 140 °C (see Fig. 4c).
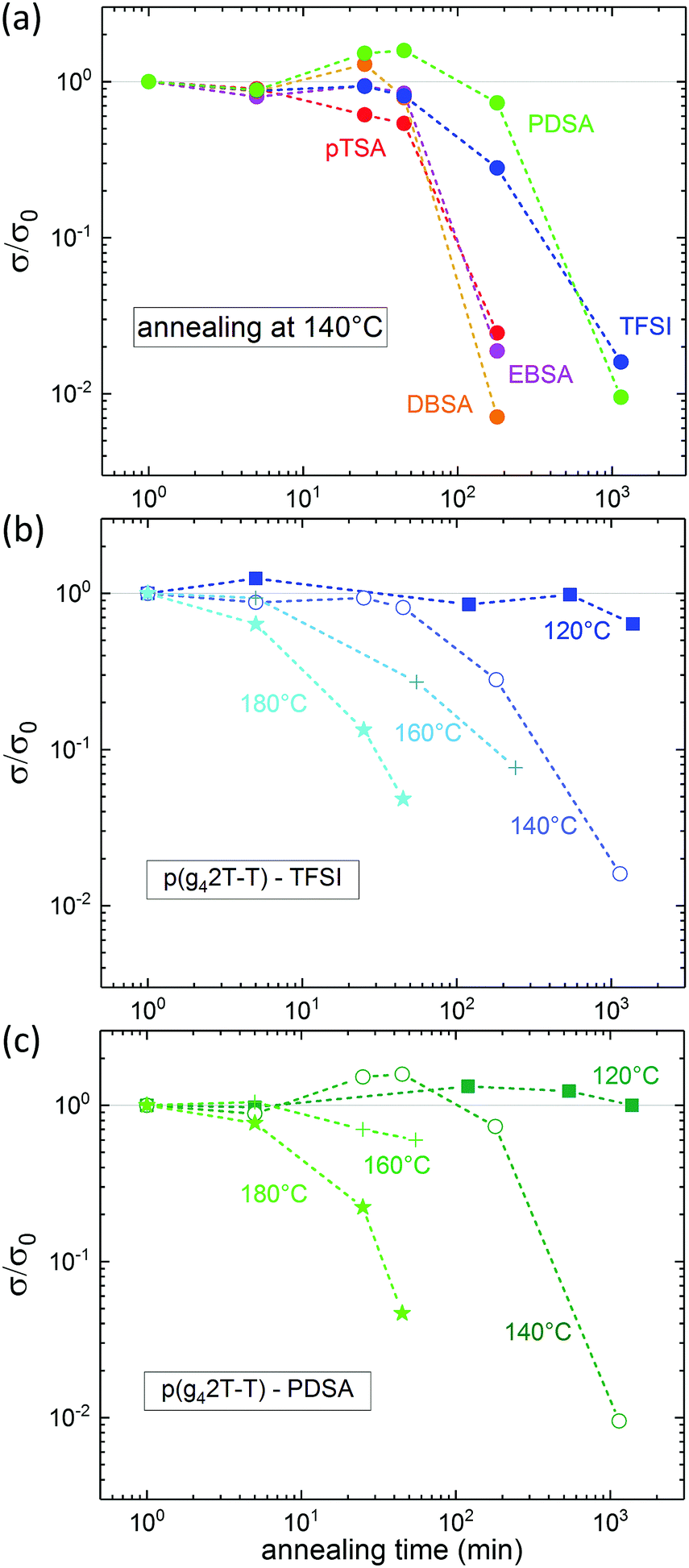 | ||
| Fig. 4 Relative conductivity as a function of annealing time (a) of p(g42T-T) doped with different acids upon annealing at 140 °C, (b) of p(g42T-T):TFSI and (c) of p(g42T-T):PDSA upon annealing at 120 °C to 180 °C. | ||
Thermogravimetric analysis (TGA) of pTSA, EBSA, DBSA and PDSA doped p(g42T-T) did not reveal any sublimation of the dopant molecule from the polymer:dopant system at temperatures below 200 °C. Thus, the observed decrease in conductivity is likely to originate from a degradation of the polymer in the presence of the strong acid (see Fig. S6, ESI†). In contrast, for p(g42T-T):TFSI we observed weight loss starting from about 100 °C onwards, which can be attributed to sublimation of excess acid dopant (boiling point of TFSI: 90 °C) and the gradual degradation of the polymer (see Fig. S6, ESI†).
The exceptional thermal stability of p(g42T-T):PDSA opens up processing routes, which are usually not viable for doped conducting polymers, such as hot pressing of bulk samples. To illustrate the potential of these alternative processing routes, we fabricated a 130 μm think film by hot pressing a mixture of solid p(g42T-T) with 25 mol% PDSA at 140 °C (see Fig. 5). The obtained freestanding, flexible film displayed an electrical conductivity of 55 S cm−1, which is equivalent to the conductivity of thin drop-casted and annealed p(g42T-T):PDSA films with the same composition.
 | ||
| Fig. 5 Free standing p(g42T-T):PDSA film fabricated by hot pressing at 140 °C. | ||
A study on the ageing of TFSI and pTSA doped p(g42T-T) revealed that the long-term stability of the electrical conductivity depends strongly on the acid concentration within the polymer (see Fig. S7, ESI†). At low acid concentrations a significant increase of the electrical conductivity was observed after six months of ageing at ambient conditions, which indicates a rise of the doping level and confirms the slow kinetics of the acid doping. For high acid concentrations the conductivity of p(g42T-T) was found to decrease over time, which suggests that an excess of acid in the polymer causes degradation. P(g42T-T) films containing an intermediate concentration of the acid dopant, however, retained a stable electrical conductivity for at least six months.
In summary, we have demonstrated that the polar polythiophene derivative p(g42T-T) can be efficiently doped by various acids using different processing routes such as sequential doping and co-processing. The most conductive films with conductivities of up to 120 S cm−1 were obtained by co-processing in the presence of oxygen and remained stable for six months under ambient conditions. Furthermore, we found that the nature of the acid dopant does not decisively influence the conductivity of the doped p(g42T-T) films, which renders acid doping a promising strategy considering the huge diversity of possible dopant molecules. The thermal stability of the doped p(g42T-T), however, was found to be strongly dependent on the acid dopant. The highest thermal stability was obtained for PDSA doped p(g42T-T), which retained its initial conductivity for 20 hours upon annealing at 120 °C and allowed hot pressing of the doped polymer into 130 μm thick, freestanding conducting films. Taking into account the high maximum conductivity of p(g42T-T):PDSA, its excellent processability from solution and in bulk, as well as the low price and toxicity of the acid dopant, the di-acid PDSA is a very promising alternative to commonly used redox dopants.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Swedish Research Council through grant no. 2016-06146, the Knut and Alice Wallenberg Foundation through a Wallenberg Academy Fellowship, and the European Research Council (ERC) under grant agreement no. 637624. The authors thank Cornell High Energy Synchrotron Source (CHESS) (supported by the NSF & NIH/NIGMS via NSF award DMR-1332208) for providing experimental time for GIWAXS measurements.References
- I. E. Jacobs and A. J. Moulé, Adv. Mater., 2017, 29, 1703063 CrossRef PubMed
.
- H. E. Katz and J. Huang, Annu. Rev. Mater. Res., 2009, 39, 71–92 CrossRef
- B. Lüssem, C.-M. Keum, D. Kasemann, B. Naab, Z. Bao and K. Leo, Chem. Rev., 2016, 116, 13714–13751 CrossRef PubMed
- F. Guillain, J. Endres, L. Bourgeois, A. Kahn, L. Vignau and G. Wantz, ACS Appl. Mater. Interfaces, 2016, 8, 9262–9267 CrossRef PubMed
- H. Yan, J. G. Manion, M. Yuan, F. P. García de Arquer, G. R. McKeown, S. Beaupré, M. Leclerc, E. H. Sargent and D. S. Seferos, Adv. Mater., 2016, 28, 6491–6496 CrossRef PubMed
- Z. Shang, T. Heumueller, R. Prasanna, G. F. Burkhard, B. D. Naab, Z. Bao, M. D. McGehee and A. Salleo, Adv. Energy Mater., 2016, 6, 1601149 CrossRef
- R. Kroon, D. A. Mengistie, D. Kiefer, J. Hynynen, J. D. Ryan, L. Yu and C. Müller, Chem. Soc. Rev., 2016, 45, 6147–6164 RSC
- B. Russ, A. Glaudell, J. J. Urban, M. L. Chabinyc and R. A. Segalman, Nat. Rev. Mater., 2016, 1, 16050 CrossRef
- Q. Zhang, Y. Sun, W. Xu and D. Zhu, Energy Environ. Sci., 2012, 5, 9639 RSC
- S. Qu, Q. Yao, L. Wang, Z. Chen, K. Xu, H. Zeng, W. Shi, T. Zhang, C. Uher and L. Chen, NPG Asia Mater., 2016, 8, e292 CrossRef
- Q. Zhang, Y. Sun, W. Xu and D. Zhu, Macromolecules, 2014, 47, 609–615 CrossRef
- D. T. Duong, C. Wang, E. Antono, M. F. Toney and A. Salleo, Org. Electron., 2013, 14, 1330–1336 CrossRef
- S. N. Patel, A. M. Glaudell, D. Kiefer and M. L. Chabinyc, ACS Macro Lett., 2016, 5, 268–272 CrossRef
- S. N. Patel, A. M. Glaudell, K. A. Peterson, E. M. Thomas, K. A. O’Hara, E. Lim and M. L. Chabinyc, Sci. Adv., 2017, 3, e1700434 CrossRef PubMed
- K. Kang, S. Watanabe, K. Broch, A. Sepe, A. Brown, I. Nasrallah, M. Nikolka, Z. Fei, M. Heeney, D. Matsumoto, K. Marumoto, H. Tanaka, S. Kuroda and H. Sirringhaus, Nat. Mater., 2016, 15, 896–902 CrossRef PubMed
- A. M. Glaudell, J. E. Cochran, S. N. Patel and M. L. Chabinyc, Adv. Energy Mater., 2015, 5, 1401072 CrossRef
- I. E. Jacobs, E. W. Aasen, J. L. Oliveira, T. N. Fonseca, J. D. Roehling, J. Li, G. Zhang, M. P. Augustine, M. Mascal and A. J. Moulé, J. Mater. Chem. C, 2016, 4, 3454–3466 RSC
- D. T. Duong, H. Phan, D. Hanifi, P. S. Jo, T. Q. Nguyen and A. Salleo, Adv.
Mater., 2014, 26, 6069–6073 CrossRef PubMed
- M. Chayer, K. Faïd and M. Leclerc, Chem. Mater., 1997, 9, 2902–2905 CrossRef
- R. H. Karlsson, A. Herland, M. Hamedi, J. A. Wigenius, A. Åslund, X. Liu, M. Fahlman, O. Inganäs and P. Konradsson, Chem. Mater., 2009, 21, 1815–1821 CrossRef
- R. Kroon, D. Kiefer, D. Stegerer, L. Yu, M. Sommer and C. Müller, Adv. Mater., 2017, 29, 1700930 CrossRef PubMed
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Müller, ACS Energy Lett., 2018, 278–285 CrossRef PubMed
- J. Liu, L. Qiu, R. Alessandri, X. Qiu, G. Portale, J. J. Dong, W. Talsma, G. Ye, A. A. Sengrian, P. C. T. Souza, M. A. Loi, R. C. Chiechi, S. J. Marrink, J. C. Hummelen and L. J. A. Koster, Adv. Mater., 2018, 30, 1704630 CrossRef PubMed
- J. Liu, L. Qiu, G. Portale, M. Koopmans, G. ten Brink, J. C. Hummelen and L. J. A. Koster, Adv. Mater., 2017, 29, 1701641 CrossRef PubMed
- L. Qiu, J. Liu, R. Alessandri, X. Qiu, M. Koopmans, R. W. A. Havenith, S. J. Marrink, R. C. Chiechi, L. J. A. Koster and J. C. Hummelen, J. Mater. Chem. A, 2017, 5, 21234–21241 RSC
- J. Li, C. W. Rochester, I. E. Jacobs, E. W. Aasen, S. Friedrich, P. Stroeve and A. Moulé, Org. Electron., 2016, 33, 23–31 CrossRef
- C. C. Han and R. L. Elsenbaumer, Synth. Met., 1989, 30, 123–131 CrossRef
- A. Trummal, A. Rummel, E. Lippmaa, I. Koppel and I. A. Koppel, J. Phys. Chem. A, 2011, 115, 6641–6645 CrossRef PubMed
- R. J. Mammone and A. G. MacDiarmid, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 105 RSC
- J. Hynynen, D. Kiefer and C. Müller, RSC Adv., 2018, 8, 1593–1599 RSC
- J. Hynynen, D. Kiefer, L. Yu, R. Kroon, R. Munir, A. Amassian, M. Kemerink and C. Müller, Macromolecules, 2017, 50, 8140–8148 CrossRef PubMed
- E. Lim, K. A. Peterson, G. M. Su and M. L. Chabinyc, Chem. Mater., 2018, 30, 998–1010 CrossRef
- D. T. Scholes, P. Y. Yee, J. R. Lindemuth, H. Kang, J. Onorato, R. Ghosh, C. K. Luscombe, F. C. Spano, S. H. Tolbert and B. J. Schwartz, Adv. Funct. Mater., 2017, 27, 1702654 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tc01593g |
| This journal is © The Royal Society of Chemistry 2018 |