
Interfacial effects on solution-sheared thin-film transistors†
 *a
*a
Abstract
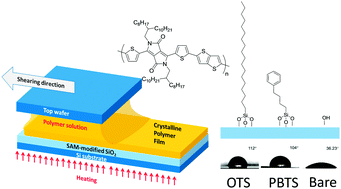
Meniscus-guided solution-sheared fabrication processes are considered a promising approach to develop high-performance polymer-based transistors owing to their enhancement of electrical performance and improvement of cystalline structure. However, the effect of the interface on the molecular packing and charge transport in the solution-sheared devices remains unclear. Therefore, in this study, we investigated interfacial effects on the electrical properties and crystalline morphology of solution-sheared polymer films based on a high-mobility donor–acceptor copolymer, poly(diketopyrrolo[3,4-c]pyrrole-co-thieno[3,2-b]thiophene) (PBDT-co-TT). We employed solution-shearing processes for making substrates modified with octadecyl trimethoxylsilane (OTS) and phenylbutyltrimethoxysilane, and bare substrates for device fabrication. The highest mobility obtained with OTS devices was 1.77 cm2 V−1 s−1, which is much higher than that obtained with the bare structure. In addition, OTS-treated devices exhibited the highest polymer alignment. According to the analysis of thicknesses and shearing speed, we conclude that OTS-modified substrates provide an ideal interface, fulfilled by simple mass balance near the meniscus, in contrast to the bare substrate. Grazing incidence X-ray diffraction analysis revealed that the OTS-based film showed the longest coherence length and a tunable lamellar d-spacing distance with shearing speeds. In contrast, the film made on the bare subtrate exhibited the smallest coherence length and a negligible change of lamellar d-spacing distance with shearing speeds. Thus, this study demonstrates the importance of the interface on polymer alignment, charge transport and meta-stable molecular packing for the solution shearing process. This may enhance the application of solution processes in the electronics industry.
- This article is part of the themed collection: Journal of Materials Chemistry C Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

