
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct and continuous hydrothermal flow synthesis of thermochromic phase pure monoclinic VO2 nanoparticles†
Delphine
Malarde‡
 a,
Ian D.
Johnson‡
a,
Ian J.
Godfrey
a,
Michael J.
Powell
a,
Giannantonio
Cibin
b,
Raul
Quesada-Cabrera
a,
Jawwad A.
Darr
a,
Claire J.
Carmalt
a,
Gopinathan
Sankar
a,
Ivan P.
Parkin
a and
Robert G.
Palgrave
*a
a,
Ian D.
Johnson‡
a,
Ian J.
Godfrey
a,
Michael J.
Powell
a,
Giannantonio
Cibin
b,
Raul
Quesada-Cabrera
a,
Jawwad A.
Darr
a,
Claire J.
Carmalt
a,
Gopinathan
Sankar
a,
Ivan P.
Parkin
a and
Robert G.
Palgrave
*a
aDepartment of Chemistry, Materials Chemistry Centre, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: r.palgrave@ucl.ac.uk
bDiamond Light Source Ltd, Diamond House Harwell Science and Innovation Campus Didcot, Oxfordshire OX11 0DE, UK
First published on 22nd October 2018
Abstract
Monoclinic vanadium(IV) oxide [VO2(M)] is a widely studied material due to its thermochromic properties and its potential use in energy-efficient glazing applications. VO2(M) nanoparticles can be a great advantage for energy-efficient glazing as below 50 nm the nanoparticles poorly interact with visible wavelengths – resulting in an increase in visible light transmittance whilst maintaining the thermochromic response of the material. The direct synthesis of VO2(M) nanoparticles with effective thermochromic properties will be a step forward towards industrial applications of this material. Unfortunately, many of the synthesis processes reported so far involve multiple steps, including post-treatment, and the synthesis is not always reproducible. In this study, we present the first direct synthesis of pure monoclinic VO2 nanoparticles by continuous hydrothermal flow synthesis (CHFS). TEM images showed that nanoparticles in the size range of 30–40 nm were produced. The VO2(M) nanoparticles also showed good thermochromic properties with a solar modulation (ΔTsol) of 3.8%, as established by UV/Vis spectroscopy. A range of analytical methods was used to characterise the materials, including X-ray absorption spectroscopy (XANES and EXAFS), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and transmission electron microscopy (TEM). The influence of niobium (Nb) doping on the physical and thermochromic properties of the VO2 nanoparticles was also explored. Previous work has shown a sharp metal-to-semiconductor transition of VO2 upon incorporation of a Nb dopant. The results of the current work suggested that these changes are likely due to changes on the local structure of the oxide.
Introduction
The numerous crystal structures and phase transitions of vanadium(IV) oxide (VO2) make it a promising material for a wide range of applications including batteries,1,2 gas sensors3 and solar control coatings for smart windows.4,5 The monoclinic form of vanadium dioxide, VO2(M) (space group P121/c1) has attracted great interest due to its thermochromic properties. VO2(M) has the ability to reversibly transform from the semiconducting monoclinic phase VO2(M) to the semi-metallic rutile phase VO2(R) (space group P42/mnm).6 The reversible metal-to-semiconductor transition (MST) occurs at ca. 68 °C for a typical undoped VO2 material.7 When heated beyond this temperature, the material's optical properties change drastically – from highly transmissive to highly reflective – in the NIR region, and its electrical conductivity increases by several orders of magnitude.8 The thermochromic properties of VO2(M) and its relatively low temperature transition, i.e. close to room temperature, make it an interesting material for energy-efficient glazing applications.The strong absorption of VO2(M), however, as well as its high reflectivity due to its narrow band gap has a negative impact on both visible light transmittance (Tlum) and solar modulation (ΔTsol) – two essential parameters for measuring the energy saving efficiency of glazing. Tlum indicates the percentage of visible light passing through the coated glass while ΔTsol represents the ability of the coating to modulate the solar energy. It had been quite challenging to optimize both parameters as the increase of Tlum often induces a reduction of ΔTsol, and vice versa.
Li et al.9,10 showed that both parameters Tlum and ΔTsol could be significantly improved simultaneously by embedding VO2 nanoparticles in a host material rather than synthesising a continuous VO2 film. It has been established, for example, that a 5 μm thick film of 1 vol% of well-dispersed VO2 nanoparticles shows higher Tlum and ΔTsol values compared to an equivalent continuous VO2 film with a thickness of 50 nm.11 In addition, it has been demonstrated that VO2 nanoparticles of ca. 50 nm (or less) weakly interact with visible light while those with particle sizes of ca. 20 nm or less suppress optical scattering.4,12 Hence, it is important to tailor particle sizes and dispersion thereof.
The direct synthesis of VO2(M) nanoparticles can be challenging due to the multiple valence states of the vanadium ion and the ease of oxidation/reduction of V4+ to V5+/V3+ species. The synthesis of phase pure monoclinic VO2 can require several steps including post-synthesis heat-treatment. Any additional processing of the material risks oxidation or reduction of the vanadium, reducing or destroying the functionality. Hydrothermal synthesis is a common route to obtain VO2(M) nanoparticles, however, this method typically requires 24 to 48 hours to produce phase pure VO2(M) nanoparticles in a batch process and it is hardly reproducible.13,14
Continuous hydrothermal flow synthesis (CHFS) is a method used to produce nanoparticles. CHFS has the advantage of being a direct, reproducible and scalable method.15 Unlike batch hydrothermal processes, CHFS is a continuous process, thus once a system is optimised it is possible to produce kg of material per hour. In CHFS, a stream of supercritical water is mixed with an ambient temperature stream of metal precursor salts (in an engineered mixer) to almost instantly form nanoparticles in a good yield (typically above 85%). In this process, metal oxide nanoparticles are produced very rapidly by the hydrolysis and subsequent dehydration (or degradation) of the metal salts.16,17 Previously, CHFS has been utilised to prepare VO2(M) and Nb-doped VO2(M) samples in a two-step process, where the initial VO2 product contained impurities and required post-synthesis annealing at 600 °C to obtain phase pure VO2(M).1,18,19 This was synthesised at a reaction mixing temperature of 335 °C using balanced flow rates for the sc-water and ambient temperature metal salt feed. However, to the authors’ knowledge, there are no reports on the successful direct synthesis of pure VO2(M) using a continuous method.
In this study, we present the first direct continuous hydrothermal flow synthesis of VO2(M) nanoparticles, which has been achieved by the modification of the CHFS apparatus and under unbalanced flow rate conditions. A vanadium(IV) precursor solution, obtained from the reduction of vanadium(V) oxide (V2O5) by oxalic acid (H2C2O4), was used for the synthesis of the nanoparticles. The phase purity and the characteristics of the nanoparticles were investigated by X-ray absorption spectroscopy (XANES and EXAFS), X-ray diffraction, X-ray photoelectron spectroscopy and transmission electron microscopy. The thermochromic properties of the as-formed nanoparticles were determined by variable temperature UV-Vis spectroscopy. This study presents a key step towards the industrial applications of thermochromic VO2 materials.
Experimental
All chemical compounds used in the synthesis were purchased from Sigma-Aldrich.Synthesis of the precursor solution
An aqueous V5+-containing solution (5 L) was prepared from a mixture of 181.88 g of V2O5 and 360.12 g oxalic acid in D.I. water (vanadium![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oxalic acid, 1:4 ratio). This solution (0.4 M) was stirred for 48 h until it was fully reduced, as evidenced by a colour change to a blue V4+-containing solution. This initial solution was diluted to 0.2 M and used directly in the CHFS process as a precursor solution.
:oxalic acid, 1:4 ratio). This solution (0.4 M) was stirred for 48 h until it was fully reduced, as evidenced by a colour change to a blue V4+-containing solution. This initial solution was diluted to 0.2 M and used directly in the CHFS process as a precursor solution.
Continuous hydrothermal flow synthesis (CHFS) of VO2 nanoparticles
VO2(M) was synthesised using a lab-scale CHFS process, which was modified from previous reports to achieve higher mixing-point temperatures.20 This required 80 mL min−1 of 450 °C D.I. water and 10 mL min−1 combined precursor feeds to obtain a theoretical mixing point temperature of ca. 402 °C.18 The hot water feed was provided by a Primeroyal K pump (Milton Roy, Pont Saint-Pierre, France), and the precursors were provided by two Gilson-type pumps (Gilson 305, Fig. 1, pumps G1 and G2) operating at flow rates of 5 mL min−1 each. The flow rates were controlled using the actuators on the pumps, which were calibrated using the LabView software (LabView, National Instruments, Berkshire, UK). Both pumps G1 and G2 provided the V4+ precursor detailed in the previous paragraph. The outputs of the pumps were connected to pressure gauges (EN 837-1, WIKA Instruments Ltd, Redhill, UK), and pressure relief valves (R3A series, Swagelok) set to a relief pressure of 276 bar, a lift check valve (50 Series, Swagelok) and a non-return valve (CH Series, Swagelok) in series. The outputs of the Gilson pumps combined in a 1/8′′ tee-piece, which subsequently flowed into a 4-way switch valve. This switch valve was also connected to the fourth pump (P4 Milton Roy Primeroyal K pump), an exhaust pipe with a Tescom BPR and the mixing point in the reactor. The 4-way valve (7921F4Y, LAA UK Ltd, Middlesex, UK) enabled the Gilson pumps to be pressurized and depressurized in isolation from the reactor, i.e. pump P4 and the Gilson pumps could be interchanged using the valve.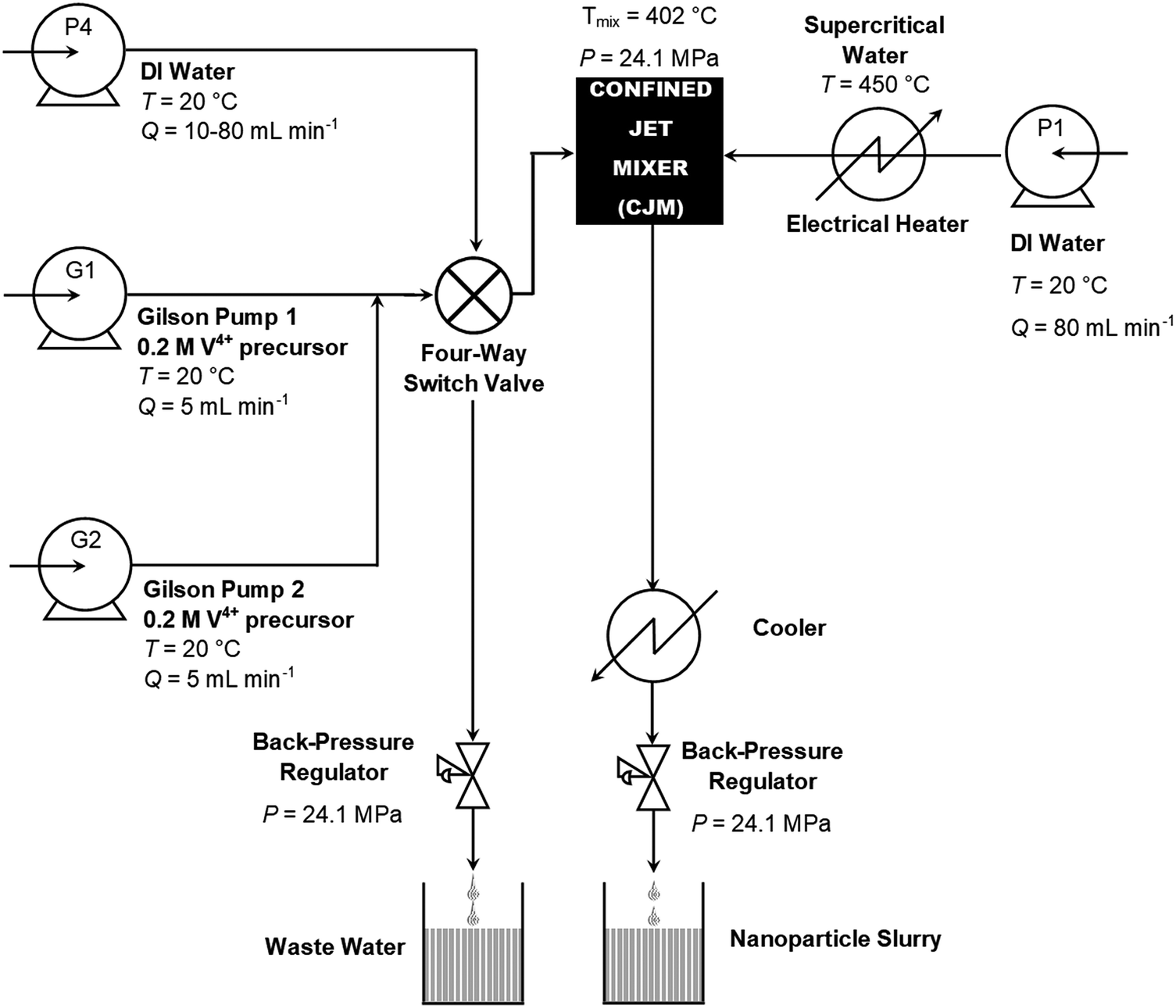 | ||
| Fig. 1 Schematic diagram of the continuous hydrothermal flow synthesis (CHFS) process used in the direct synthesis of VO2(M) nanoparticles. | ||
During synthesis, G1 and G2 were both employed to feed the 0.2 M V4+ precursor solution into the reactor. The combined output of G1 and G2 flowed into a Confined Jet Mixer (CJM), where it combined with the hot water feed from P1.21 The theoretical mixing temperature was 402 °C for all reactions. The Reynolds number of the combined feed was ca. 14000 and 22000 for the 1/8′′ and 1/16′′ inlet pipe reactor configurations, namely configurations A and B, respectively. Configuration A was employed to synthesise the pure VO2 sample while configuration B was used to synthesise the Nb-doped VO2 sample. In both cases, the extremely high Reynolds numbers resulted in very rapid and turbulent mixing between the hot water feed and the precursors. Thus, the CJM allowed complete mixing within 50 ms of combination,21 which occurred just beyond the supercritical water inlet pipe within the cross-piece as indicated in Fig. S1 (ESI†). The extremely high Reynolds number resulted in very rapid and turbulent mixing between the hot water feed and the precursors, which therefore caused rapid nucleation of the VO2 product within the 1/4′′ outlet pipe of the CJM (Fig. S1, ESI†). As the temperature of the combined feeds was 402 °C, the water solvent was still in the supercritical state. The output slurry flowed through a 1.4 m section of the 1/4′′ pipe, giving a residence time of 1.0 s, before flowing through a pipe-in-pipe counter-current cooling section to reduce the slurry temperature to ca. 40 °C. The cooled product slurry exited the apparatus via a back-pressure regulator (Tescom, model 26-1762-24-194, Emerson Process Management Regulator Technologies Inc., South Lanarkshire, UK), which maintained a pressure of 240 bar in the system.
After synthesis, the supernatant was decanted from the product slurry and the concentrated paste was centrifuged at 4500 rpm for 5 minutes. The centrifuged sludge was repeatedly resuspended in D.I. water and centrifuged until the conductivity of the supernatant was reduced below 100 μS m−1. The cleaned, wet product was placed in a freeze-drier (Virtis Genesis 35XL) and slowly heated from −60 °C to 25 °C, over 24 h under a vacuum of <13 Pa.
Film analysis
X-Ray Diffraction (XRD) was performed using a Stoe StadiP diffractometer in the transmission mode (coupled θ–2θ geometry), with the sample sandwiched between two plastic foil disks held together with a thin layer of silicone grease. A pre-sample Ge(111) monochromator selected Mo-Kα1 radiation only (λ = 0.709 Å) and included a 0.5 mm collimator restricted to 3 mm height. The sample was rotated in the beam, and the diffraction intensity was recorded using a Dectris Mythen 1k silicon strip detector covering 18° (2θ). XRD patterns were collected in the 2θ range of 2° to 40° with a step size of 0.02° in 2θ and were compared to the ICSD reference pattern for monoclinic VO2 (ICSD 34033).The surface morphologies of the films were determined by Scanning Electron Microscopy (SEM) using a JEOL JSM-6700 (3 kV).
X-ray photoelectron spectroscopy (XPS) was carried out on powder using a Thermo Scientific K-alpha spectrometer with monochromated Al Kα radiation, a dual beam charge compensation system and at a constant pass energy of 50 eV (spot size 400 μm). Spectra were recorded from 0 to 1200 eV.
Transmission electron microscopy (TEM) images were obtained using a TEM Jeol 2100 with a LaB6 source operating at an acceleration voltage of 200 kV.
The optical transmittances were monitored using a Perkin Elmer Lambda 950 UV-Vis-NIR spectrophotometer that was equipped with a homemade heating unit. The powder was deposited onto a silica barrier (50 nm SiO2 layer) float glass, supplied by Pilkington/NSG. The thermochromic properties of the deposited films were measured by recording their transmittance spectra from 2500 nm to 250 nm at two different temperatures (20 °C and 90 °C).
To investigate the local structure of the samples, V K-edge X-ray absorption spectra were recorded on beamline B18 at diamond light source.22 B18 was equipped with a Si(111) dual-crystal monochromator and Pt-coated harmonic rejection mirrors. The samples were pressed into pellets, with cellulose added as the binding agent, and data were recorded in transmission mode; data for a V foil reference were recorded simultaneously with each sample. Commercial monoclinic vanadium dioxide was obtained from Sigma Aldrich and measured as a standard compound. Multiple spectra of each sample were averaged prior to data analysis. Data reduction and fitting were carried out using the Demeter 0.9.25 suite of programmes.23 Data fitting was performed in R-space.
Results and discussion
Continuous hydrothermal flow synthesis and characterisation
Continuous hydrothermal flow synthesis (CHFS) was used to produce VO2(M) nanoparticles in a single step as a black powder suspension, which was cleaned and freeze-dried to yield a black powder. The vanadium(IV) precursor solution used for the synthesis was obtained from the reduction of vanadium(V) oxide by oxalic acid according to the accepted decomposition pathway shown in eqn (R1)–(R3)18,24,25| V2O5(s) + 4H2C2O4 + 2H2O → [(VO)2(C2O4)3]2− + 2H3O+ + 2CO2 + 4H2O | (R1) |
| [(VO)2(C2O4)3]2− + 2H3O+ → 2VOC2O4 + CO + CO2 + 3H2O | (R2) |
| 2VOC2O4 → VO2 + CO + CO2 | (R3) |
The X-ray diffraction (XRD) patterns of the undoped and Nb-doped samples both matched the VO2(M) reference exclusively (PDF Card No. 01-076-0456) (Fig. 2). An additional broad feature observed in these patterns at 2θ ∼ 5° was due to the silicone-based matrix used during the sample preparation.26 This is in direct contrast to previous CHFS works, which resulted in a mixture of VO2 phases and required heat treatment to 600 °C to produce the phase-pure VO2(M).18 It is suggested that the higher reaction temperature (ca. 402 °C compared to 335 °C) employed herein was necessary to drive complete conversion to the VO2(M) phase in flow. The ability to manufacture VO2(M) directly in a continuous process has no precedent, as the majority of synthesis methods are batch hydrothermal, and require long reaction times in the order of days.27,28 Furthermore, the precursors are relatively benign (oxalic acid is commonly used to remove rust) in comparison to these other methods (which often require chemicals with significant hazards such as hydrazine).
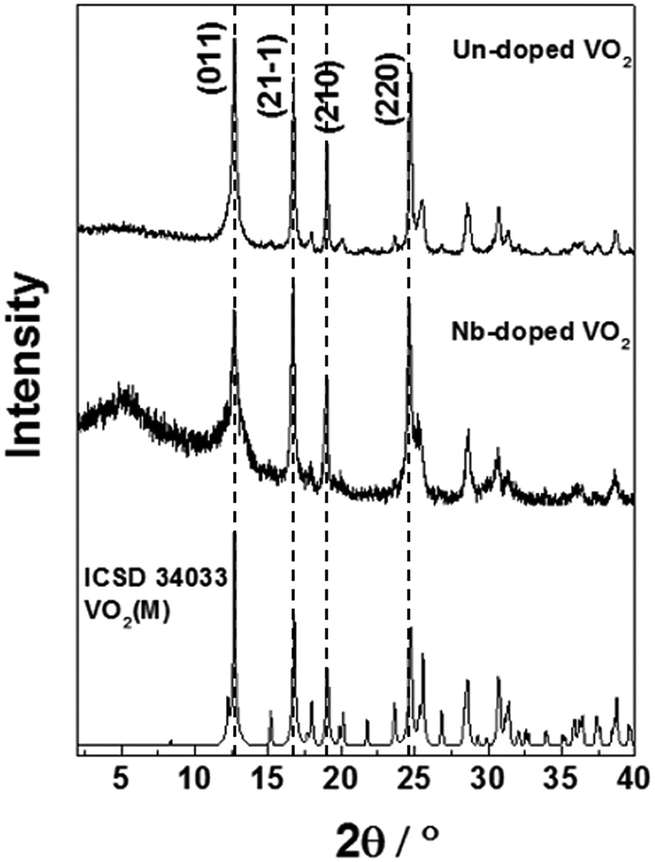 | ||
| Fig. 2 XRD patterns of the VO2(M) and Nb-VO2(M) nanoparticles synthesised by CHFS. The standard XRD pattern of monoclinic VO2 (ICSD 34033) has been included for reference. | ||
The undoped VO2 sample showed high crystallinity with relatively narrow peak broadening, as estimated from the FWHM values (Table 1a). In comparison with the literature pattern, a preferential orientation along the (2 2 0) plane could be observed for our samples, which is indicated by an increase in the intensity of the (2 2 0) peak compared with the (0 1 1) and (2 1 ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) peaks. Peak broadening is slightly higher in the case of the Nb-doped VO2 sample. This suggests the Nb dopant caused the reduced crystallinity, as the particle sizes of both samples were similar and therefore broadening due to the particle size effects should be approximately equal. We observe a slight change in the relative intensities of the diffraction peaks upon incorporation of the Nb dopant, which could be due to changes in the degree of preferred orientation of the Nb doped samples, or difference in the electron density between Nb5+ and V4+. The crystal lattice parameters and the monoclinic angles of the VO2 and Nb-VO2 samples, as estimated from the refinement of the XRD patterns, are summarized in Table 1. They correspond to the monoclinic M2 phase, where VO2 crystallizes in a centered monoclinic system with the space group C2/m.29 VO2 with the M2 phase has been previously reported in the literature for doped VO2 systems and VO2 materials obtained from stress-induced processes.29,30 This phase has attracted interest in Mott transition studies.
) peaks. Peak broadening is slightly higher in the case of the Nb-doped VO2 sample. This suggests the Nb dopant caused the reduced crystallinity, as the particle sizes of both samples were similar and therefore broadening due to the particle size effects should be approximately equal. We observe a slight change in the relative intensities of the diffraction peaks upon incorporation of the Nb dopant, which could be due to changes in the degree of preferred orientation of the Nb doped samples, or difference in the electron density between Nb5+ and V4+. The crystal lattice parameters and the monoclinic angles of the VO2 and Nb-VO2 samples, as estimated from the refinement of the XRD patterns, are summarized in Table 1. They correspond to the monoclinic M2 phase, where VO2 crystallizes in a centered monoclinic system with the space group C2/m.29 VO2 with the M2 phase has been previously reported in the literature for doped VO2 systems and VO2 materials obtained from stress-induced processes.29,30 This phase has attracted interest in Mott transition studies.
| Samples | Crystal lattice parameters (Å) | Angle (°) | Cell volume V | ||
|---|---|---|---|---|---|
| a | b | c | β | ||
| VO2(M) | 9.133(1) | 5.674(1) | 4.495(1) | 90.69(2) | 232.92(6) |
| Nb-VO2(M) | 9.088(2) | 5.784(1) | 4.514(1) | 91.47(1) | 237.3(1) |
SEM analysis (Fig. 3) showed round-shaped particles for both VO2 and Nb-VO2 samples. This morphology differs from the VO2 nanoparticles previously synthesised by CHFS, which showed rod-like particles.18 This could suggest that the higher reaction temperature used in this study (402 °C) promoted rapid nucleation that resulted in the rounded nanoparticles. It can also be pointed out that the high uniformity of the sample indicated the ability of the CHFS process to produce consistent quality nanoparticles in a short time.
 | ||
| Fig. 3 SEM images of the un-doped VO2(M) powder. | ||
TEM analysis of the undoped VO2 and Nb-doped VO2 samples revealed both samples were formed of a mixture of spherical particles and hexagonal particles (Fig. 4a and b). The particle size analysis of the VO2(M) and Nb-VO2(M) samples (50 particles per study) revealed average particle sizes of 33 ± 13 nm and 39 ± 13 nm, respectively (Fig. S2, ESI†). The small particle sizes observed in general were likely due to the high Reynolds number (i.e. more turbulent, rapid mixing) for both samples. The smaller particle sizes achieved herein compare extremely favourably with those made previously via CHFS, where the particle sizes were in the range of 50–200 nm and particle sizes below 50 nm were only achieved upon significant Nb dopant inclusion. Moreover, the post-synthesis heat-treatment required in the previous study could significantly increase the particle sizes and cause the risk of oxidation. Herein, the direct synthesis of monoclinic VO2 nanoparticles with smaller particle sizes makes this ideal for thermochromic solar control coatings. Lattice fringes, which could be assigned to the VO2(M) lattice spacings, were observed in both cases (Fig. 4c and d). In addition, an organic surface coating could be observed on some particles (Fig. 4a), which visibly degraded in the beam. It is suggested that surplus oxalic acid is present in the synthesis bound to the surface of the VO2 particles.
 | ||
| Fig. 4 TEM images of (a) undoped VO2 and (b) Nb-doped VO2. Lattice fringes of (c) undoped VO2 and (d) Nb-doped VO2. | ||
The oxidation states of vanadium and niobium were determined by X-ray photoelectron spectroscopy (XPS). The V 2p3/2 core-level peak of the undoped sample could be modelled and fitted with two environments, as shown in Fig. 5. It showed the presence of two oxidation states of vanadium, V4+ and V5+, at binding energies of 514.8 eV and 516.3 eV, respectively. This indicated that the VO2 powder is partially oxidized to V2O5 at the extreme surface. This is due to the powder being stored in air at room temperature and forming a V2O5 passivation layer. The same observation applied to the Nb-doped powder sample. The XPS spectra are shown in Fig. 5. The atomic percentage of the Nb dopant in the doped sample was estimated to be <1 at% by modelling the Nb 3d5/2 core-level peak. The Nb 3d5/2 core-level peak is observed at a binding energy of 207.0 eV, which corresponds to the oxidation state of the Nb5+ ion. The low intensity of the Nb 3d signal makes definitive chemical identification impossible, so it cannot be ruled out that other chemical environments of Nb are present. Nb doping did not have any significant effect on the oxidation of the VO2 powder as both samples have been shown to have the same V4+:V5+ ratio (ca. 33:67). This is in contrast to the previous study, where significant changes in the V4+:V5+ ratios were observed upon Nb incorporation – resulting in an increase of V4+ ions in the doped sample.19 All the above binding energies are in agreement with literature values (±0.2 eV).31,32
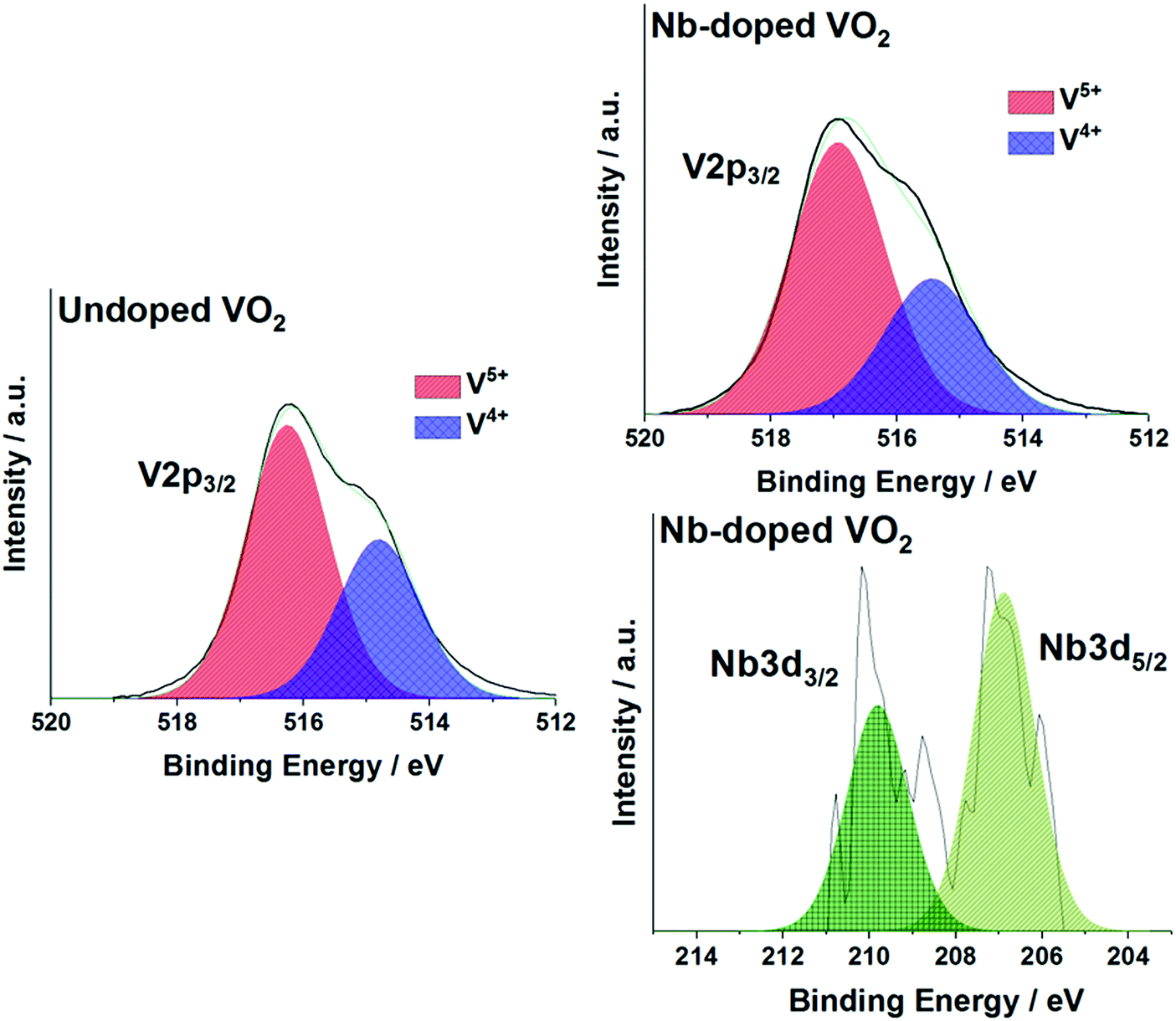 | ||
| Fig. 5 X-ray photoelectron spectra (XPS) of the V 2p3/2 and Nb 3d5/2 environments in the (a) undoped VO2 and (b) Nb-VO2 samples. | ||
The low levels of Nb detected make the influence of the dopant on the samples questionable. The low atomic percentage could be due to the precursor solution used during the synthesis. Piccirillo et al.33 showed that in the synthesis of Nb-VO2 films by chemical vapour deposition methods, a Nb/V molar ratio threshold of ca. 7.5% was found in order to detect Nb incorporation into the films. This had also been observed with APCVD processes.34 In the current study, the precursor solution contained a Nb/V molar ratio of 1.25% and this could explain the low dopant level in the powder. In addition, no changes in the V4+:V5+ ratio and the particle sizes were observed between the undoped and Nb-doped samples unlike the previous study – suggesting that there is only a small amount of Nb incorporated.19 Nevertheless, the XRD patterns showed some evidence of substitution of the V4+ atoms by the Nb5+ atoms.
X-ray absorption spectroscopy
V K-edge X-ray absorption near-edge structure (XANES) spectra for these samples are shown in Fig. 6. The samples are all close in appearance to the monoclinic VO2 standard, confirming that they are phase-pure.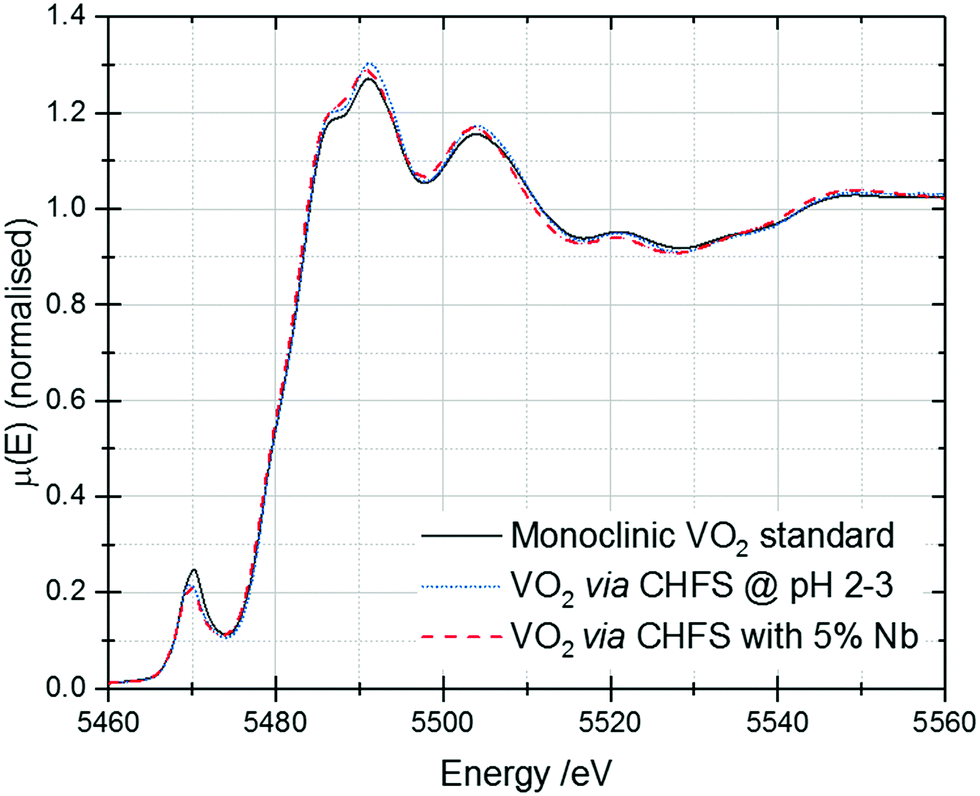 | ||
| Fig. 6 V K-edge XANES of the VO2 and Nb-VO2 samples synthesised by CHFS. A spectrum of a commercial monoclinic VO2 standard has been included for reference. | ||
V K-edge extended X-ray absorption fine structure (EXAFS)/Fourier transforms and best fits are shown in Fig. 7. These data were fitted to a theoretical structure based on ICSD 34033;35 the fitting model used was similar to that reported by Marini et al.36 The best fit parameters from these fits are given in Table 2. It should be noted that different k-space windows were used for the Fourier transforms for the different samples – this is responsible for the differences in the appearance of the samples in Fig. 7a. This was done to include the maximum possible data range in the fit, while excluding noise/high-k artefacts – this enables fitting of the highest possible number of parameters to the best precision.
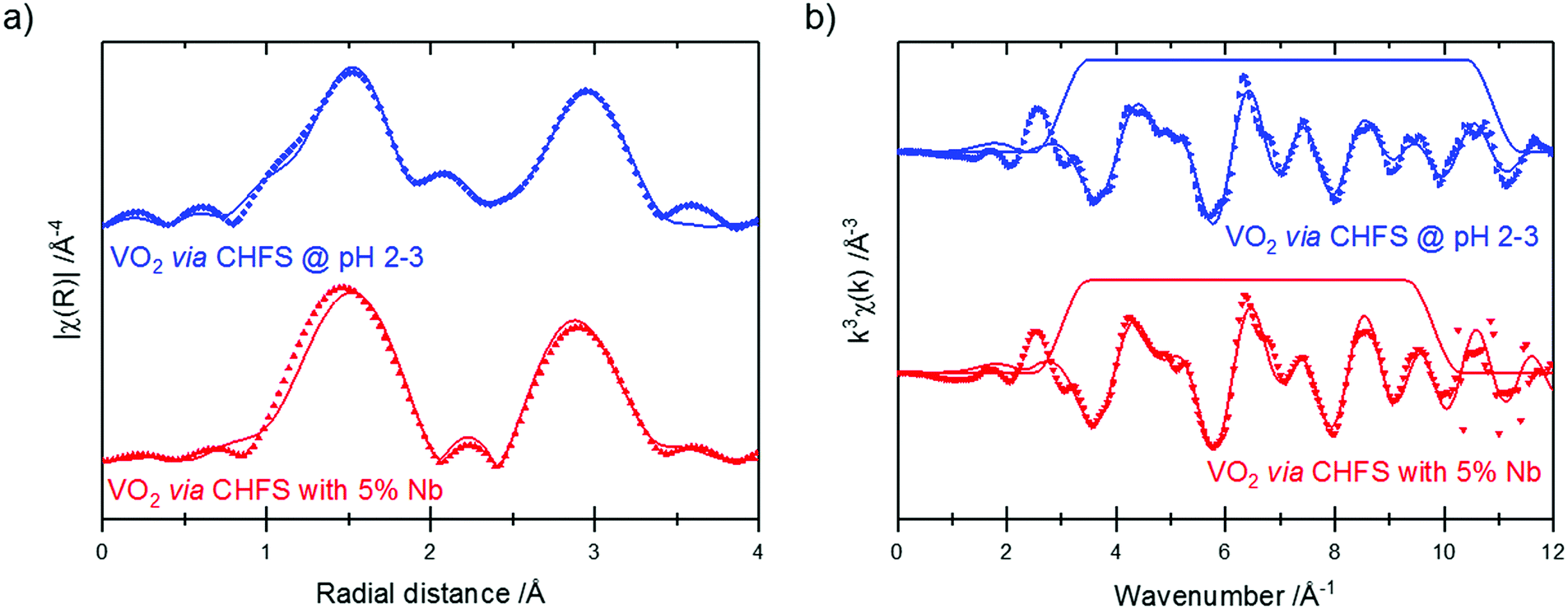 | ||
| Fig. 7 V K-edge Fourier transforms (a) and EXAFS (b) of the CHFS VO2 samples. Data are plotted as symbols, and fits as solid lines. The trapezoid lines in (b) indicate the Fourier transform window used. | ||
| Sample | Path | Degeneracy | R/Å | σ 2/Å−2 | R factor |
|---|---|---|---|---|---|
| VO2via CHFS | V–O | 1 | 1.83(5) | 0.009(5) | 0.008 |
| 2 | 1.96(2) | ||||
| 2 | 2.37(4)δ | 0.016(9)η | |||
| 1 | 2.42(4)δ | ||||
| V–V | 1 | 2.61(3) | 0.008(4) | ||
| 1 | 3.17(9) | ||||
| 2 | 3.41(9)ε | ||||
| 2 | 3.49(9)ε | ||||
| 2 | 3.4(2)ζ | 0.016(9)η | |||
| 2 | 3.5(2)ζ | ||||
| Nb-VO2via CHFS | V–O | 1 | 1.85(1)θ | 0.0025(9)λ | 0.010 |
| 2 | 1.97(1)θ | ||||
| 2 | 2.41(7)ι | 0.013(4) | |||
| 1 | 2.46(7)ι | ||||
| V–V | 1 | 2.7(2) | 0.02(1)μ | ||
| 1 | 3.3(4) | ||||
| 2 | 3.42(2)κ | 0.0025(9)λ | |||
| 2 | 3.50(2)κ | ||||
| 2 | 3.59(2)κ | 0.013(4)μ | |||
| 2 | 3.66(2)κ |
The EXAFS of each of the samples were broadly consistent with the literature structure of monoclinic vanadium dioxide; however, the two longest V–O distances showed a degree of expansion (without Nb: 2.37 ± 0.04 Å, 2.42 ± 0.04 Å; with Nb: 2.41 ± 0.077 Å, 2.46 ± 0.07 Å; literature: 2.02 Å, 2.06 Å), while all the other distances were (within uncertainty) in line with the literature structure. An increase in (some of) the V–O distances while the V–V distances remain constant suggests that there is an out-of-plane displacement of the oxygen ions, as indicated by the purple arrows in Fig. 8.
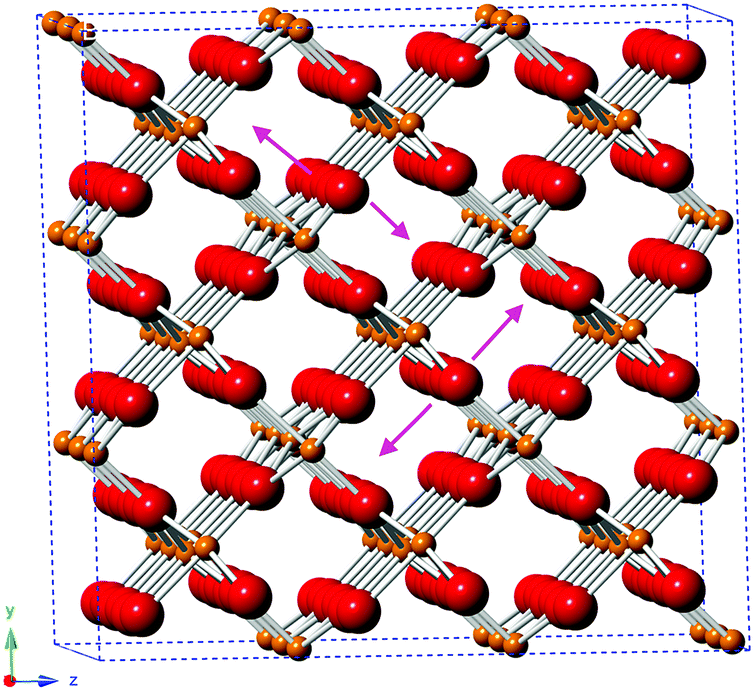 | ||
| Fig. 8 Structure of monoclinic VO2, based on ICSD 34033, indicating the direction of the proposed out-of-plane oxygen displacement (purple arrows). O2−: red; V4+: orange. | ||
The longer V–O paths, which show the greatest expansion, also show increased Debye–Waller factors, indicating increased disorder in these paths. The increased disorder in the out-of-plane direction indicated in Fig. 8 would explain both the increased Debye–Waller factors and the increased average path length in this direction.
The Nb-doped sample showed a slight increase in the average distance for these V–O bonds. This suggests that the substitution of Nb5+ into the vanadium dioxide lattice slightly increases this disorder, although this cannot be established with certainty due to the uncertainties on the fitted parameters.
Thus, the EXAFS data confirmed the monoclinic structure of these vanadium dioxide samples and confirmed that this structure is maintained after Nb doping. It also identified out-of-plane disorder of some of the oxygen atoms.
Thermochromic properties
The optical properties of the VO2(M) nanoparticles were measured by recording their transmittance spectra from 250 nm to 2500 nm up to 90 °C (Fig. 9). All samples displayed thermochromic behaviour with a reversible switch around ca. 68 °C – corresponding to the typical transition temperature expected for undoped VO2. The luminous and solar transmittance (Tlum and Tsol) were estimated based on the measured spectra using the equation below (Table 3):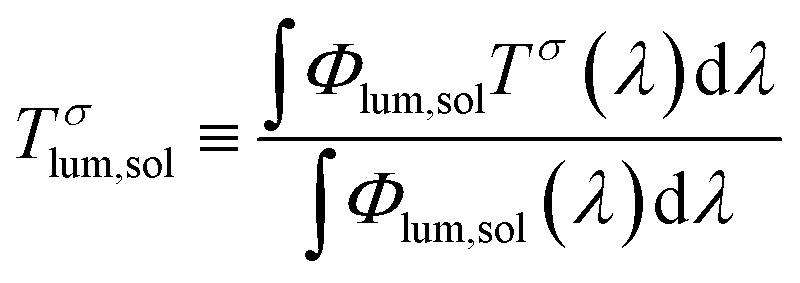 | (1) |
| ΔTsol = Tsol (25 °C) − Tsol (90 °C) | (2) |
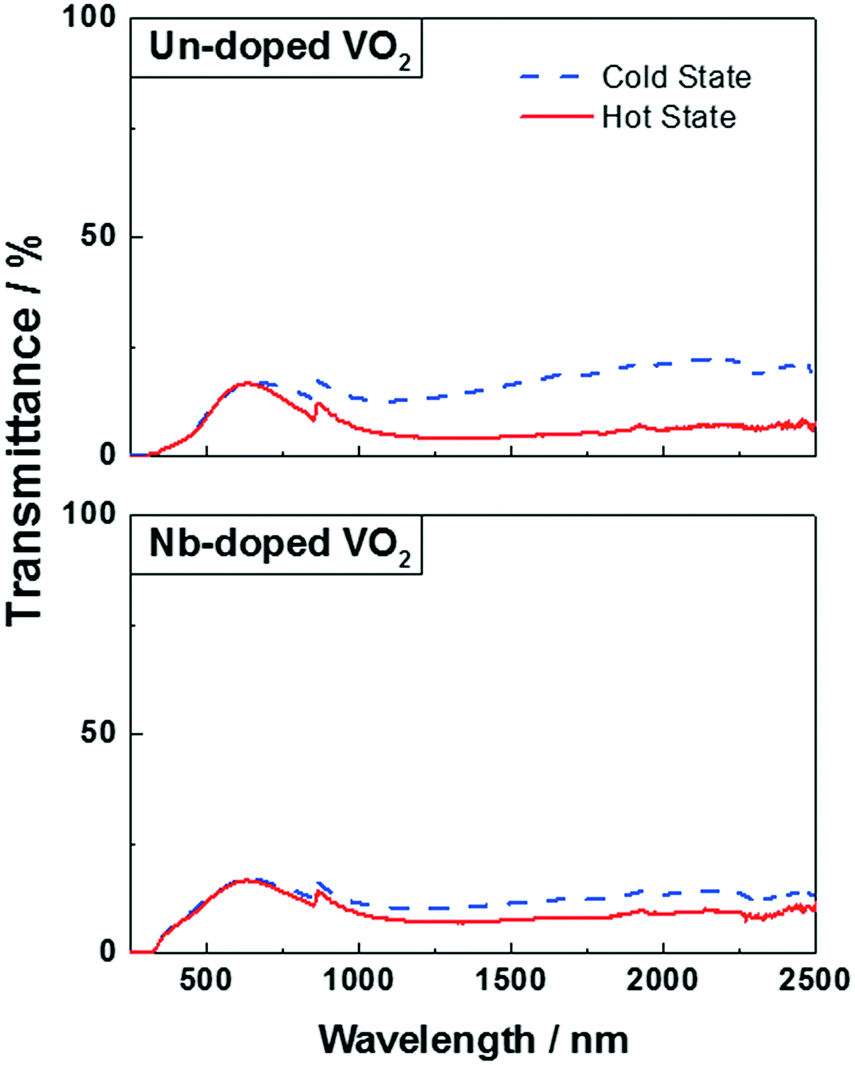 | ||
| Fig. 9 UV-Vis spectra of un-doped VO2 (top) and Nb-doped VO2 (bottom) at 20 °C (blue dashed line) and 90 °C (continuous red line). | ||
| Samples | T lum (%) | ΔTsol (%) | ΔTIR (%) | |
|---|---|---|---|---|
| Cold state | Hot state | |||
| Un-doped VO2 | 14 | 14 | 3.8 | 7 |
| Nb-doped VO2 | 15 | 15 | 1.5 | 2.4 |
The modulation in the infrared region (ΔTIR) was also estimated from eqn (2) as it only takes into account the near-infrared region (750 to 2500 nm) of the measured spectra, which is specific for the thermochromic functionality expected.
The undoped VO2 powder showed a ΔTsol of 3.8%, which is in line with those of the un-doped VO2 materials synthesised by other methods such as magnetron sputtering or APCVD.37–39 The powder exhibited a good efficiency for modulating infrared radiation with a ΔTIR of 7%, however, it displayed a low visible light transmittance (Tlum = 14%).
Several studies showed that doping VO2 samples with niobium is usually accompanied by a decrease of the visible light transmittance Tlum and a MST reduction.40–42 Both samples, however, have a similar Tlum of 15% and 14% for the doped and un-doped samples, respectively. Moreover, the transition temperature (MST) of the doped sample is around ca. 68 °C, which is the MST observed for a typical undoped VO2 material. This can be explained by the very low at% of niobium present in the powder – the atomic percentage is too low for observing any effect on the optical properties. On the other hand, Table 3 shows a poor ability of the Nb-doped sample to modulate infrared radiation, which can be seen by a drop in the ΔTsol (1.5%) and ΔTIR (2.4%) when compared to the undoped powder. This is most likely due to the lower crystallinity of the doped material. It has been shown that a poor crystallinity of the VO2 samples resulted in a low solar modulation.39 Furthermore, both samples have been shown to have the same V4+:V5+ ratio and similar average particle sizes.
As a result, the undoped VO2 nanoparticles synthesised by CHFS showed good thermochromic properties. Both samples have been revealed to have a low luminous transmittance, which could be caused by the sample preparation for UV-Vis measurements. The small average particle sizes of the samples suggest that a good dispersion of the NPs in a host could significantly increase the Tlum. Doping with niobium did not seem to have any impact on the luminous transmittance and the MST. Only a change in the ΔTsol has been observed and is most likely a result of the differing degrees of crystallinity.
Conclusions
Pure monoclinic VO2 nanoparticles (undoped and doped with niobium) were successfully synthesised by CHFS at a mixer reaction temperature of ca. 400 °C. The synthesis was direct and no post-synthesis heat-treatment of the samples was required. The vanadium(IV) precursor solution was immediately converted into a VO2(M) nanoparticle slurry in the process. The undoped VO2 powder possessed good thermochromic properties, a ΔTsol of 3.8% and a ΔTIR of 7%, but a low visible light transmittance, which could be attributed to the sample preparation. The Nb-doped sample was synthesised using a Nb/V solution with a molar ratio of 1.25%. The XRD patterns and XANES and EXAFS analyses highlighted some evidence of Nb5+ substitutions into the VO2 lattice, suggesting that the Nb dopant induced a relatively poor VO2 crystallinity. Nevertheless, the molar ratio of the Nb/V solution was too low to have any significant effect on the MST and the optical properties of the sample, or for definitive conclusions on the nature of the Nb in the sample to be ascertained. Despite the low luminous transmittance for both samples, the TEM images showed small particle sizes (in the range of 30 to 40 nm), which could lead to a significant increase of the Tlum if the particles are well dispersed in a host in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by the chemistry department of the UCL and the Engineering and Physical Sciences Research Council, EPSRC (EP/M003353/1 intelligent coatings for energy efficient glazing, ICE glazing). Diamond is acknowledged for access to B18 under proposal SP17147-1. IDJ is funded via the JUICED energy hub (EP/R023662/1). CJC and MJP acknowledge EPSRC grant EP/L017709/1.References
- M. Lübke, N. Ding, M. J. Powell, D. J. L. Brett, P. R. Shearing, Z. Liu and J. A. Darr, Electrochem. Commun., 2016, 64, 56–60 CrossRef.
- L. Jiang, Y. Qu, Z. Ren, P. Yu, D. Zhao, W. Zhou, L. Wang and H. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 1595–1601 CrossRef CAS PubMed.
- Y. Qin, G. Fan, K. Liu and M. Hu, Sens. Actuators, B, 2014, 190, 141–148 CrossRef CAS.
- J. Zhou, Y. Gao, Z. Zhang, H. Luo, C. Cao, Z. Chen, L. Dai and X. Liu, Sci. Rep., 2013, 24, 1–5 Search PubMed.
- G. Sun, X. Cao, X. Gao, S. Long, M. Liang and P. Jin, Appl. Phys. Lett., 2016, 109, 143903 CrossRef.
- Z. Zhang, Y. Gao, Z. Chen, J. Du, C. Cao, L. Kang and H. Luo, Langmuir, 2010, 26, 10738–10744 CrossRef CAS PubMed.
- C. G. Granqvist, S. Green, G. A. Niklasson, N. R. Mlyuka, S. von Kræmer and P. Georén, Thin Solid Films, 2010, 518, 3046–3053 CrossRef CAS.
- D. Vernardou, M. E. Pemble and D. W. Sheel, Chem. Vap. Deposition, 2006, 12, 263–274 CrossRef CAS.
- S. Y. Li, G. A. Niklasson and C. G. Granqvist, J. Appl. Phys., 2011, 109, 1–6 Search PubMed.
- S. Y. Li, G. A. Niklasson and C. G. Granqvist, J. Appl. Phys., 2010, 108, 63525 CrossRef.
- C. G. Granqvist, Mater Today: Proc., 2016, 3, S2–S11 CrossRef.
- K. Laaksonen, S. Y. Li, S. R. Puisto, N. K. J. Rostedt, T. Ala-Nissila, C. G. Granqvist, R. M. Nieminen and G. A. Niklasson, Sol. Energy Mater. Sol. Cells, 2014, 130, 132–137 CrossRef CAS.
- N. Wang, Q. Sheng, P. Lin, S. Magdassi and Y. Long, J. Alloys Compd., 2017, 711, 222–228 CrossRef CAS.
- J. Zhu, A. Huang, K. Tong, S. Ji, S. Bao, X. Cao and P. Jin, ACS Appl. Mater. Interfaces, 2016, 8, 29742–29748 CrossRef CAS PubMed.
- J. A. Darr, J. Zhang, N. M. Makwana and X. Weng, Chem. Rev., 2017, 117, 11125–11238 CrossRef CAS PubMed.
- M. Lübke, I. Johnson, N. M. Makwana, D. Brett, P. Shearing, Z. Liu and J. A. Darr, J. Power Sources, 2015, 294, 94–102 CrossRef.
- T. Adschiri, K. Kanazawa and K. Arai, J. Am. Ceram. Soc., 1992, 22, 1019–1022 CrossRef.
- M. J. Powell, P. Marchand, C. J. Denis, J. C. Bear, J. A. Darr and I. P. Parkin, Nanoscale, 2015, 7, 18686–18693 RSC.
- R. Quesada-Cabrera, M. J. Powell, P. Marchand, C. J. Denis, J. A. Maggio, F. Di Darr and I. P. Parkin, J. Nanosci. Nanotechnol., 2016, 16, 10104–10111 CrossRef CAS.
- R. I. Gruar, C. J. Tighe and J. A. Darr, Ind. Eng. Chem. Res., 2013, 52, 5270–5281 CrossRef CAS.
- I. D. Johnson, M. Loveridge, R. Bhagat and J. A. Darr, ACS Comb. Sci., 2016, 18, 665–672 CrossRef CAS PubMed.
- A. J. Dent, G. Cibin, S. Ramos, A. D. Smith, S. M. Scott, L. Varandas, R. M. Pearson, N. A. Krumpa, C. P. Jones and P. E. Robbins, J. Phys. Conf. Ser., 2009, 190, 12039 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- D. N. Sathyanarayana and C. C. Patel, J. Inorg. Nucl. Chem., 1965, 27, 297–302 CrossRef CAS.
- D. N. Sathyanarayana and C. C. Patel, J. Inorg. Nucl. Chem., 1966, 28, 2277–2283 CrossRef CAS.
- T. G. Ros, M. K. Van Der Lee, A. J. Van Dillen, J. W. Geus and D. C. Koningsberger, J. Mol. Catal. A: Chem., 2002, 186, 13–24 CrossRef CAS.
- J. H. Son, J. Wei, D. Cobden, G. Cao and Y. Xia, Chem. Mater., 2010, 22, 3043–3050 CrossRef CAS.
- J. Zhou, Y. Gao, X. Liu, Z. Chen, L. Dai, C. Cao, H. Luo, M. Kanahira, C. Sun and L. Yan, Phys. Chem. Chem. Phys., 2013, 15, 7505 RSC.
- M. Marezio, D. B. McWhan, J. P. Remeika and P. D. Dernier, Phys. Rev. B: Solid State, 1972, 5, 2541–2551 CrossRef.
- K. Okimura, T. Watanabe and J. Sakai, J. Appl. Phys., 2012, 111, 1–7 CrossRef.
- G. Silversmit, D. Depla, H. Poelman, G. B. Marin and R. De Gryse, J. Electron Spectrosc. Relat. Phenom., 2004, 135, 167–175 CrossRef CAS.
- P. E. Sobol and J. Chastain, Handbook of X-ray Photoelectron Spectroscopy, 1993 Search PubMed.
- C. Piccirillo, R. Binions and I. P. Parkin, Eur. J. Inorg. Chem., 2007, 4050–4055 CrossRef CAS.
- T. D. Manning, I. P. Parkin, M. E. Pemble, D. Sheel and D. Vernardou, Chem. Mater., 2004, 16, 744–749 CrossRef CAS.
- J. M. Longo and P. Kierkegaard, Acta Chem. Scand., 1970, 24, 420–426 CrossRef CAS.
- C. Marini, S. Pascarelli, O. Mathon, B. Joseph, L. Malavasi and P. Postorino, EPL, 2013, 102, 66004 CrossRef.
- N. R. Mlyuka, G. A. Niklasson and C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2009, 93, 1685–1687 CrossRef CAS.
- M. J. Powell, R. Quesada-Cabrera, A. Taylor, D. Teixeira, I. Papakonstantinou, R. G. Palgrave, G. Sankar and I. P. Parkin, Chem. Mater., 2016, 28, 1369–1376 CrossRef CAS.
- D. Malarde, M. J. Powell, R. Quesada-cabrera, R. L. Wilson, C. J. Carmalt, G. Sankar, I. P. Parkin and R. G. Palgrave, ACS Omega, 2017, 2, 1040–1046 CrossRef CAS.
- Y. Li, Y. Liu, J. Liu and L. Ren, J. Mater. Sci.: Mater. Electron., 2016, 27, 4981–4987 CrossRef CAS.
- H. M. Pinto, J. Correia, R. Binions, C. Piccirillo, I. P. Parkin and V. Teixeira, Mater. Sci. Forum, 2008, 587, 640–644 Search PubMed.
- C. Batista, J. Carneiro, R. M. Ribeiro and V. Teixeira, J. Nanosci. Nanotechnol., 2011, 11, 9042–9045 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tc00859k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |