
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular design principles for efficient photoresponsive polymer–azobenzene complexes
Jaana
Vapaavuori
 *ab,
C. Geraldine
Bazuin
*b and
Arri
Priimagi
*a
*ab,
C. Geraldine
Bazuin
*b and
Arri
Priimagi
*a
aLaboratory of Chemistry and Bioengineering, Tampere University of Technology, P.O. Box 541, FI-33101 Tampere, Finland. E-mail: arri.priimagi@tut.fi
bDépartment de Chimie, Université de Montréal, C.P. 6128, Succursale Centre-Ville, Montréal, QC H3C 3J7E, Canada. E-mail: jaana.vapaavuori@umontreal.ca; geraldine.bazuin@umontreal.ca
First published on 23rd January 2018
Abstract
Noncovalent binding of azobenzenes to polymers allows harnessing light-induced molecular-level motions (photoisomerization) for inducing macroscopic effects, including photocontrol over molecular alignment and self-assembly of block copolymer nanostructures, and photoinduced surface patterning of polymeric thin films. In the last 10 years, a growing body of literature has proven the utility of supramolecular materials design for establishing structure–property–function guidelines for photoresponsive azobenzene-based polymeric materials. In general, the bond type and strength, engineered by the choice of the polymer and the azobenzene, influence the photophysical properties and the optical response of the material system. Herein, we review this progress, and critically assess the advantages and disadvantages of the three most commonly used supramolecular design strategies: hydrogen, halogen and ionic bonding. The ease and versatility of the design of these photoresponsive materials makes a compelling case for a paradigm shift from covalently-functionalized side-chain polymers to supramolecular polymer–azobenzene complexes.
 Jaana Vapaavuori | Jaana Vapaavuori defended her doctoral thesis, “The Supramolecular Design of Efficient Photoresponsive Materials”, done under supervision of Dr Arri Priimagi and Prof. Matti Kaivola, in the department of Applied Physics at Aalto University, Helsinki, Finland, in 2013. Currently, she is a Banting Postdoctoral Fellow in the Department of Chemistry at the University of Montreal, Canada working together with Prof. Geraldine Bazuin and Prof. Christian Pellerin. Her main research interests involve developing new materials and design concepts for light-controllable nanostructures and liquid crystals, for all-optical surface patterning of polymers, and for electrochemical devices. |
 C. Geraldine Bazuin | Geraldine Bazuin, who obtained her PhD at McGill University in 1984 on the subject of ionomers, followed by a postdoc in Strasbourg, France in the area of liquid crystals, was professor first at Université Laval from 1986 to 2003, then at Université de Montréal since 2003. She led one of the pioneering research groups working with supramolecular side-chain liquid crystal polymers, focusing in particular on ionically bonded systems. Recently, this work has included azo-containing small molecules and has embraced (H-bonded) supramolecular block copolymer systems, particularly in the form of dip-coated and Langmuir–Blodgett thin films. |
 Arri Priimagi | Arri Priimagi earned his PhD degree in 2009 from the Department of Applied Physics, Helsinki University of Technology (presently Aalto University), Finland. After a two-year postdoc at Chemical Resources Laboratory, Tokyo Institute of Technology, and a 10-month International Fellowship at Politecnico di Milano, he was in 2014 appointed to the position of a tenure-track assistant professor at the Department of Chemistry and Bioengineering at Tampere University of Technology, Finland, where he presently works as an associate professor. In 2014 he received an Academy of Finland research Fellowship, and in 2016 was awarded an European Research Council Starting Grant entitled “Tunable Photonic Structures via Photomechanical Actuation”. His research interests lie in functional soft matter, with a particular emphasis in photonic and light-responsive systems. |
1. Introduction
1.1. Supramolecular functionalization
Design and fabrication of functional materials based on covalent chemistry has been an important topic in synthetic chemistry for more than a century. In contrast, it is only a few decades ago that supramolecular chemistry, which exploits noncovalent bonds, was established as a distinct field by Jean-Marie Lehn.1–4 Despite being a relatively young field, supramolecular chemistry has become a very successful tool and has given rise to many subfields, such as molecular self-assembly, molecular recognition, template-directed synthesis, mechanically interlocked molecular architectures, biomimetics, and molecular machinery.5 In its simplest expression, two molecules having complementary interaction sites at mutually accessible locations join together non-covalently to form a supramolecule.5 The variety and strengths of feasible supramolecular interactions is vast, ranging from strong ionic bonds with interaction strengths as high as 350 kJ mol−1 to very weak hydrogen bonds with strengths less than 5 kJ mol−1. Hence, depending on the strength of the interaction, a spectrum of different material properties can be obtained, where the weaker bonds can give an equilibrium of bonded and non-bonded states and the strongest ones have covalent-like character.6,7In the present review, focusing on recent developments in supramolecular photoresponsive materials, the latter are fabricated through ionic, hydrogen and halogen bonding between generally photopassive host materials and photoactive azobenzene units. The ionic bond consists of a coulombic attraction between two oppositely charged molecular entities.8 Hydrogen bonding refers to an electrostatic attraction between a hydrogen atom that is covalently attached to an electronegative atom (“proton donor”) and another electronegative atom (“proton acceptor”).9 Analogously, a halogen bond forms when there is a net attractive interaction between a nucleophile and a halogen atom covalently attached to an electrophilic entity.10 Hydrogen and halogen bonds allow for convenient control of the polymer–chromophore interactions by proper choice of the bond donor group, thus providing an attractive tool for studying the effect of the strength/nature of the supramolecular interaction on the photoresponsive properties of the material.11,12
The supramolecular functionalization strategy is flexible, facile and cost-effective, simplifying the preparation of libraries that isolate particular molecular parameters, such as the chromophore dipole moment, while keeping other parameters, such as polymer backbone molecular weight and polydispersity index, constant. Furthermore, it can be applied to a variety of host architectures, including linear, star-like and dendritic homopolymers and block copolymers, having the tendency to form self-assembled periodic patterns at the scale of 10–100 nm,13 the formation of which can be controlled by block-selective attachment of azobenzenes.
1.2. Azobenzene at work: photoinduced anisotropy and surface patterning
Azobenzene derivatives have been popular building blocks in photoresponsive supramolecular materials due to their clean photoisomerization reaction, consisting of a photoinduced reversible switching of the azobenzene geometry between the rod-like trans isomer and the globular, metastable cis isomer, illustrated in Fig. 1a. The ease with which various azobenzene derivatives can be synthesized is advantageous for controlling the photochemistry, which is highly dependent on the substitution pattern of the azo molecules, as amply reviewed by Bandara and Burdette14 as well as by Bleger and Hecht.15 In addition, trans azobenzene is an effective mesogenic core, utilized for instance in designing side-chain liquid crystalline polymers.16–19 Kato et al. demonstrated already in 1996 that azobenzene photoswitching can be used for driving a nematic–isotropic phase transition in a hydrogen-bonded liquid crystalline polymer–azobenzene complex.18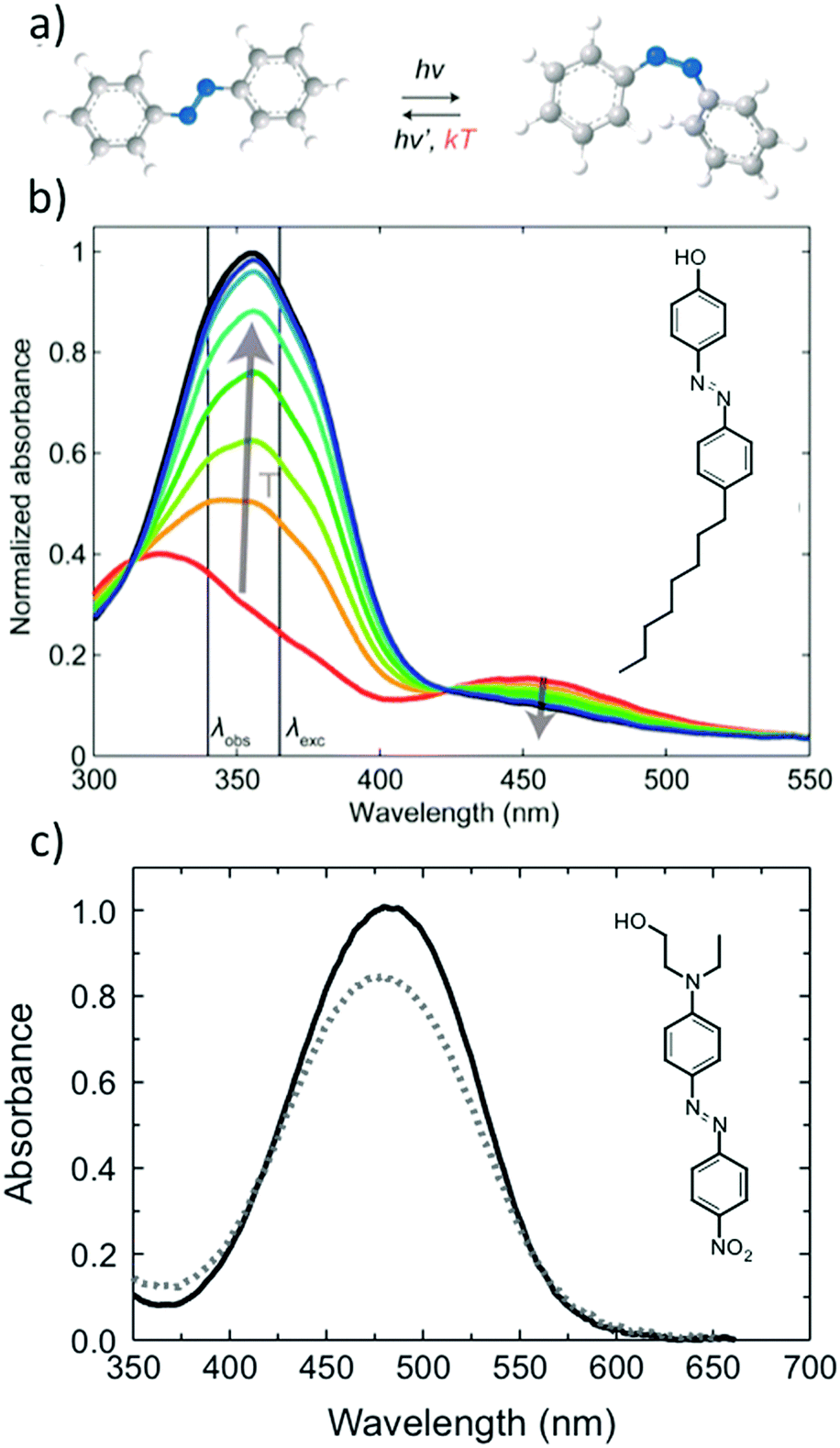 | ||
| Fig. 1 (a) Azobenzene photoisomerization reaction. The reaction from the metastable cis to the stable trans form can occur thermally or photochemically. (b) Typical example of spectral changes in a polymer–azobenzene complex, for which the azobenzene trans and cis bands are well isolated, which can give rise to bistable photoswitchable systems. The black and red curves denote the spectra in the dark and in the photostationary state, respectively, while the intermediate ones can be used for determining the thermal half-life of the cis isomer. (c) Disperse Red 1 (DR1) doped in a polymer host serves as an example of an azobenzene for which the trans and cis bands overlap, and the same (visible) wavelength drives the isomerization reaction to both directions. Fig. 1a and b adapted with permission.20 Copyright 2016, American Society of Chemistry. Fig. 1c adapted with permission.21 Copyright 2010, Optical Society of America. | ||
Upon illumination, an azobenzene-containing sample will reach a photostationary state between the photoinduced and thermally driven reactions. Depending on the pumping wavelength and the chemical structure of the azobenzene derivative in question, either trans–cis isomerization, cis–trans isomerization, or continuous cycling between the two isomers will predominantly occur. Quasi-bistable photoswitching, as illustrated in the spectra of Fig. 1b, and continuous cycling between the trans and cis isomers, as illustrated in the spectra of Fig. 1c, manifest themselves rather differently in UV-vis spectra and can bring about distinct functions, both useful in their own respect. This review will mostly concentrate on phenomena driven by the latter, being essential in photoinduced motions in amorphous azobenzene-containing polymers.
The general mechanism of photo-orientation is illustrated in the upper panel of Fig. 2. To a good approximation, the transition dipole moment of the trans azobenzene typically coincides with an axis connecting the midpoints of the two benzene rings,22 rendering the molecules that are perpendicular to the polarization plane of the incident light inert to the incident irradiation. Linearly polarized light thus orients the molecules perpendicular to the polarization plane, and the resulting anisotropy can be quantified by measuring at least three different interdependent figures of merit![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichroic ratio, order parameter, and photoinduced birefringence. The first observation of light-induced anisotropy in azobenzene-containing materials dates back to 1929, when Weigert published his pioneering work,23 giving rise to the term Weigert effect, for photo-orientation. The field was reborn in the 1970s and 1980s, with publications on photo-orientation in viscous azobenzene solutions,24,25 mixtures of polymers and azobenzene molecules26 and covalent azobenzene-functionalized polymers.27,28 Inspired by these early developments, reviewed in 198929 and in 1993,30 Natansohn and Rochon successfully developed structure–property relationship rules for amorphous, covalently-functionalized side-chain azobenzene polymers,31,32 collected in their 2002 review.33
:dichroic ratio, order parameter, and photoinduced birefringence. The first observation of light-induced anisotropy in azobenzene-containing materials dates back to 1929, when Weigert published his pioneering work,23 giving rise to the term Weigert effect, for photo-orientation. The field was reborn in the 1970s and 1980s, with publications on photo-orientation in viscous azobenzene solutions,24,25 mixtures of polymers and azobenzene molecules26 and covalent azobenzene-functionalized polymers.27,28 Inspired by these early developments, reviewed in 198929 and in 1993,30 Natansohn and Rochon successfully developed structure–property relationship rules for amorphous, covalently-functionalized side-chain azobenzene polymers,31,32 collected in their 2002 review.33
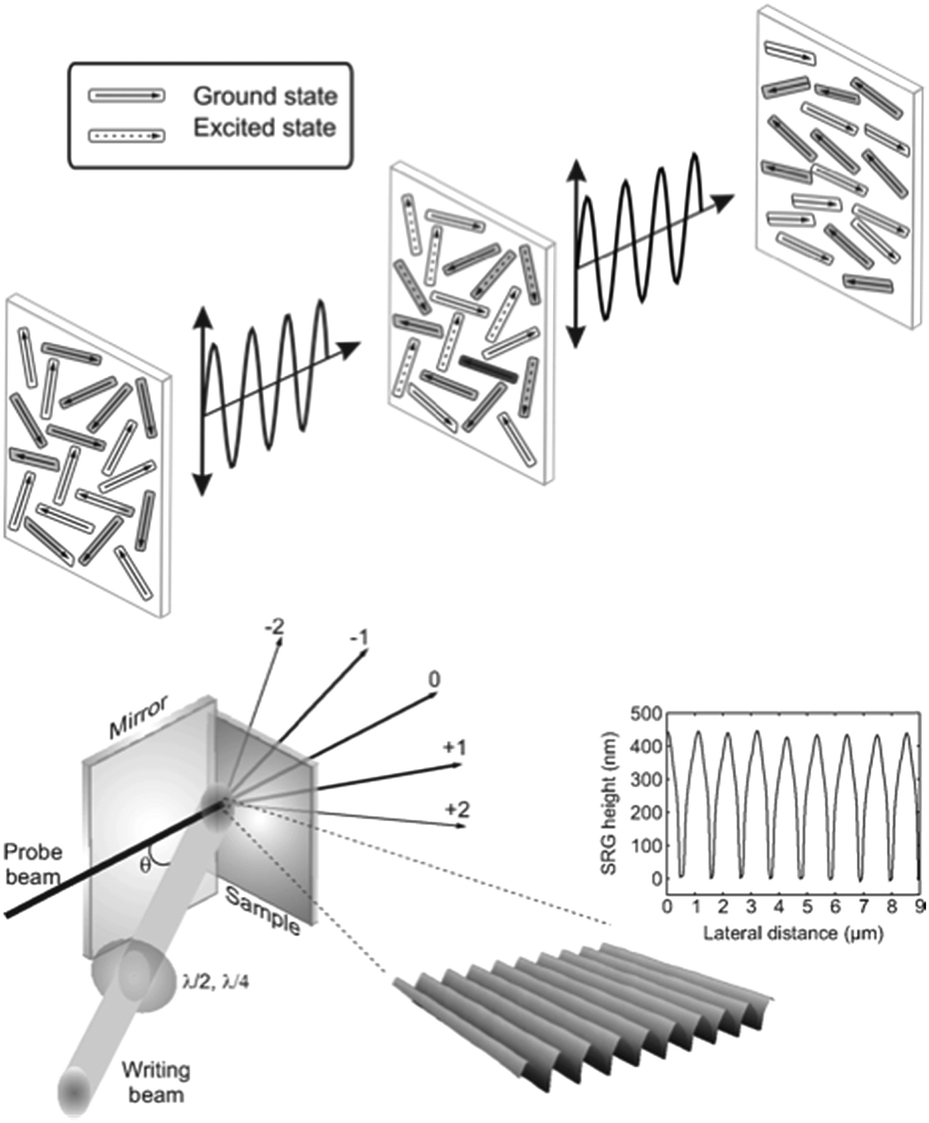 | ||
| Fig. 2 Schematic of photo-orientation in an azobenzene-containing film when illuminated with linearly polarized light (top) and surface-relief grating formation when illuminated with a polarization/intensity interference pattern (bottom). Adapted with permission.36 Copyright 2010, World Scientific Publishing. | ||
A particularly fascinating consequence of photoisomerization is polymeric mass transport under interference irradiation, over a scale of several micrometers, three orders of magnitude larger than the size of the photoisomerizing units. This all-optical surface patterning, often times denoted as surface-relief grating (SRG) formation, is schematically illustrated in the lower panel of Fig. 2 and was first observed in 1995.34,35 It can be detected in situ by monitoring light diffraction from the area of the emerging surface pattern. Information on the patterns and thus mass transport can also be gathered ex situ by nanomicroscopy techniques such as atomic force microscopy (AFM).
Despite extensive theoretical research efforts,37–40 the origin of the SRG inscription still remains a conundrum. As Ambrosio et al. and Fabbri et al. point out, it is likely that a combination of several microscopic mechanisms is necessary to describe all of the observed phenomena.41,42 Additionally, Seki and coworkers concluded that the SRG formation mechanism in low-Tg liquid-crystalline polymer films, where all of the azobenzene molecules were pre-driven into a cis state, is different from that in glassy amorphous polymer films, where the SRG formation is based on continuous cycling of the azobenzenes between the trans and cis states.43–45
Thus, it is likely that the illumination conditions and the type of material in question affect the mechanisms underlying different photoinduced patterning processes. Possibly more than one mechanism may be in effect simultaneously and/or subsequently at different phases of the surface pattern formation. Furthermore, the photo-orientation of the azobenzene molecules perpendicular to the polarization plane of the incident light occurs simultaneously with photoinduced surface patterning, leading to birefringence gratings. Interference patterns may also create other periodical changes in the material making the final diffraction grating a superposition of several gratings with different time dynamics and relative phases.46,47
1.3. Scope of this review and complementary reading
Research on organic film-forming photoresponsive materials goes back to at least 1970, with the 1980s and 1990s being the golden age of research on covalently functionalized photoresponsive azobenzene-containing polymers.30,33 To learn more about the earlier work on organic photoresponsive materials based on covalent functionalization, we refer the reader to the reviews by Viswanathan et al.,48 Natansohn and Rochon,33 and Shibaev and coworkers.49 Oliveira et al.50 and Advincula51 have summarized the progress on photoresponsive layer-by-layer thin films, not covered in the present review. In the 2000s, the focus in material design has shifted towards supramolecular analogues of these polymers, typically composed of photopassive polymers functionalized non-covalently with azobenzene molecules. A review of these materials covering the pre-2009 period has been published by Stumpe and coworkers.52 Many more papers on this subject have been published since that review, with a number of structure–property–function relations elucidated. Here, we review the progress made since 2009, focusing on the material design concepts for fabricating efficient photoresponsive materials through ionic, hydrogen and halogen bonding between the host polymers and small molecules.In 2014, Faul summarized the use of ionic self-assembly for supramolecular materials, briefly including photoresponsive azobenzene complexes,53 Ube and Ikeda reviewed recent developments in photomobile azobenzene-based materials,54 and Seki reviewed mainly covalent photoresponsive liquid crystal polymers,44 complemented by a review and a highlight article by Yu.55,56 In 2013, Priimagi et al. wrote an account on using halogen bonding in the design of functional materials57 and, in 2015, they reviewed halogen bonding in the context of photoresponsive azobenzene-containing materials.58 The interplay of light and azobenzene-containing block copolymer morphologies in solution and in the solid-state was reviewed by Zhao and He in 2009.59 Very recently, in 2017, a review of light-triggered topographies in azopolymers including a brief description of the supramolecular work in the field was written by Broer and coworkers.60 Also in 2017, the use of azobenzene as a building block in molecular machine architectures61 and in light-powered molecular devices62 were reviewed. Additionally, in early 2018 light control of nano-objects especially focusing on using noncovalent interactions to couple azobenzene surfactants to these objects was reviewed, thus indicating the timeliness of employing supramolecular interactions in the design of photoresponsive materials.63
For technological applications of light-responsive materials, which is out of the scope of the present review, we refer the readers to the reviews by Ichimura on the use of azobenzenes as LC alignment surfaces,64 Lee et al. on photofluidization lithography,65 Seki et al. on recent advancements in photoalignment,66 Natansohn and Rochon on the application potential of both photo-orientation and photo-induced surface patterning,67 and Priimagi and Schevchenko on the potential photonic applications of the photo-induced surface patterns.68
2. The extent and the stability of the supramolecular bond
In this section, we will first indicate the principal methodologies used to characterize the existence and extent of supramolecular bonding, considering both direct methods and how the presence of bonding can be evaluated by indirect methods, particularly based on material property changes. Furthermore, the methodology for in situ measurements of the bond stability during photoisomerization is discussed. Finally, the influence of the supramolecular bond strength versus competitive intermolecular interactions on the dispersion of the photoactive guest within the passive host is discussed.For pre-experimental estimates of the likelihood and strength of formation of a hypothesized supramolecular bond, energy minimization of the complex by density functional theory (DFT) is often applied, even though care should be taken to involve relevant functionals and corrections. Moreover, DFT does not provide information on which of the competing interactions of the material system in question are the dominating ones. To include all relevant interactions in one simulated system, a more informative platform is molecular dynamics (MD), since it is a many-body type simulation by nature. Regarding experimental characterization, it is imperative to consider that sample preparation conditions can have significant effects on the extent of the anticipated supramolecular bonding. For example, spin-coating leads to rapid solvent evaporation, which can freeze in non-equilibrium supramolecular structures. This may, for instance, lead to a higher extent of hydrogen bonding and therefore less phase separation (if this is a tendency), as compared to drop-casting or dip/blade-coating, for which slower solvent evaporation allows dry-state equilibrium structures to be approached or attained. In structured systems, such as block copolymers or liquid-crystalline (LC) materials, the degree of development of the morphologies formed also depends on the sample preparation conditions. For the former, heat and solvent annealing are commonly used to develop or control microphase separation, and the photoresponse of the system may depend on the degree of self-assembly of the material.69–72 On the other hand, annealing after photoinduced surface patterning has been shown, under certain conditions, to enhance pre-inscribed gratings for amphiphilic block copolymers.73
2.1. Characterization of the supramolecular bond
Demonstrating the formation and measuring the strength of existing supramolecular bonds experimentally depends strongly on the type of supramolecular bond anticipated and on the nature (amorphous vs. organized) of the final material system. Recently, dynamic force microscopy (DFM) has been used to map changes in electron-density in two-dimensional hydrogen-bonded assemblies of naphthalene tetracarboxylic diimide molecules,74 and in general, the advancement of high-resolution microscopy can further enhance the capacity to directly observe supramolecular bonds. However, typical experimental methods used for the characterization of supramolecular bonds in bulk samples can be divided into direct methods that explicitly probe the characteristics/existence of the supramolecular bonds (in an ensemble) and indirect methods that rely on the changes in material properties induced by supramolecular bonding. For ionic interactions, two categories must be distinguished: those resulting from proton transfer (often also involving some H-bonding), such as from a sulfonic acid group to pyridine, or from carboxylic acid to a tertiary amine, and those implicating ion exchange procedures between two salts.The most common experimental method for characterizing hydrogen-bonding, halogen-bonding and ionic bonding resulting from proton transfer is Fourier transform infrared spectroscopy (FTIR),75 the use of which in the context of azobenzene-materials has been reviewed recently.76 It provides information on which chemical groups are involved in the supramolecular bonding and the IR wavenumber shifts upon complexation can be qualitatively correlated with the supramolecular interaction strength.75 Additionally, quantification of the extent of supramolecular bonding can be reached by using appropriate model compounds.77 More onerous methods, like X-ray photoelectron spectroscopy (XPS) and solid-state NMR can also provide information on halogen and hydrogen bonding.78–80 For ionic bonds resulting from ion exchange, XPS and FTIR are generally not very helpful, since the changes are often subtle. Instead, the bonding is often deduced from the absence of the small counterions targeted for elimination (to drive the desired interaction), which can be measured by energy dispersive X-ray spectroscopy (EDS).
For any of the supramolecular bonds, additional confirmation of complexation can be obtained by verifying the molar ratios of the components using NMR spectroscopy and elemental analysis. This is particularly useful when excess small molecule has been introduced to obtain complete complexation and that is subsequently eliminated during ion exchange and purification (often by dialysis), and when drastic drying conditions are employed with less strongly bonded small molecules to eliminate residual solvent (e.g. under prolonged vacuum and/or high temperature).
Furthermore, changes in various properties of the supramolecular complexes compared to the parent components provide an indirect measure of the success of supramolecular bonding. Typically, the photoactive small molecules in their pure form are crystalline, whereas the complexes with a host polymer are non-crystalline. Thus, the absence of crystallinity – easily detected by wide-angle X-ray diffraction (WAXD) and complemented with differential scanning calorimetry (DSC) to confirm the absence of/change in a melting point – is supportive of complete complexation, whereas its presence is indicative of (partial) phase separation of uncomplexed material. Similarly, changes in the polymer glass transition, as detected by DSC, particularly as a function of the small molecule content, can be correlated with supramolecular bonding. In various systems, successful complexation generates liquid crystallinity, often not present in the pure components and which is generally characterized by a combination of DSC, polarized optical microscopy (POM) and X-ray techniques (WAXD and small angle scattering, which can also be applied to thin films in grazing incidence angle configuration).81–83
The combination of illumination with any of the characterization tools mentioned above can provide valuable insights into what happens to the supramolecular bond during photoisomerization, i.e. when there is photoinduced motion in the material. FTIR is the easiest method to combine with incident illumination to gain such information in situ. For example, our studies indicate that phenolic hydrogen bond and iodoacetylene-based halogen bond with poly(4-vinylpyridine) (P4VP) are relatively stable upon trans–cis–trans cycling.84 Such studies, assessing the photostabilities of different supramolecular bonds, should be extended, for instance, to different temperatures. Yet already the first experiments85 highlight that supramolecular bonds are effective for kinetic energy transfer between the phototoactive azobenzene units and the photopassive polymer.
2.2. Interplay of different interactions in supramolecular material systems
In supramolecular materials design, it is essential to take a holistic view, since the material components interact with one another not only via site-specific supramolecular interactions, but also via non-specific dispersion and electrostatic forces. Therefore, in addition to the choice of supramolecular interaction type, the choice of the chromophore itself is important, since its shape and dipolar character will determine the intermolecular interactions among the chromophores themselves. Another aspect is the chromophore-to-host ratio, which can be varied in order to tune the properties of the resulting material. These combined factors determine whether the chromophores are well dispersed within the polymer host, whether they self-assemble into ordered structures, or form macrophase-separated, often times crystalline, domains.Overall, three main possibilities can be distinguished: (i) chromophore–chromophore interactions dominate the chromophore–host interactions, leading to macrophase separation of the chromophore domains; (ii) chromophore–host interactions are strong enough to prevent macrophase separation, but lateral interactions between the chromophores lead to self-organization of the material, particularly into LC domains; and (iii) chromophore–host interactions are strong enough and/or chromophore–chromophore interactions are weak enough to maintain both supramolecular bonding and dispersion of the chromophores, resulting in a macroscopically isotropic, amorphous material even at nominally equimolar chromophore concentration. In practice, different systems (or preparation conditions) may lie at different points on a continuum outlined by these three cases.
Case (i), in which chromophore–chromophore interactions dominate over other interactions, leads to aggregated structures similar to those of the pure molecules, which are typically crystalline. Crystalline aggregates can, in fact, be useful for specific applications. For instance, Disperse Red 1 (DR1) molecules, which show a large blue-shift in the absorption maximum when they are aggregated compared to when they are dispersed, may lead to highly stable photoinduced anisotropy,21 and such systems have been proposed as humidity-controlled memory materials.85 On the other hand, H-type and J-type aggregation of azobenzenes can be used for fluorescence emission enhancement.86–88 More specifically, the Yu group found that fluorescence measurements can be employed as an indicator of aggregate size and tightness, and that the aggregates in solution state can be disassembled by photoisomerizing the azobenzenes with UV light.87,88 However, as a rule of thumb for solid-state polymer–azobenzene complexes, azobenzene aggregation, i.e. case (i), is detrimental to both the isomerization reaction and the photoisomerization-induced phenomena.89,90 Case (ii), which often results in LC character above a system-specific chromophore/host ratio, has been observed both for H-bonded16,91 and ionically bonded systems.81,92,93 Case (iii) is more likely for chromophores with low dipolar character and/or a bulkier or less linear molecular structure,94 as well as for materials with low chromophore/host ratio.
3. Supramolecular polymer–azobenzene complexes as photoresponsive materials
In the context of photoresponsive azobenzene-based materials, the interest in adopting supramolecular strategies can be related to three main advantages. In comparison to nonbonded guest–host mixtures, these strategies can prevent or decrease the aggregation of the azobenzene molecules, thus enhancing the photoresponse of the materials.89,90,95 In comparison to covalently functionalized side-chain polymers, an important advantage concerns the ease of preparation and customization of supramolecular materials, rendering them a more cost-effective and less labor-intensive choice. This is especially valuable for extensive fundamental structure–property relationship studies. Finally, the dynamic character of many noncovalent bonds can be taken advantage of by, for instance, non-destructive removal of the azo molecules using selective solvation.96,97 This chapter is divided into three topical areas: photoinduced anisotropy, photoinduced surface patterning, and photocontrol of block copolymer micro- and nanostructures, in each of which the use of supramolecular materials design has seen significant progress within the past years.3.1. Photoinduced anisotropy
As early examples of photoinduced anisotropy in supramolecular complexes, Medvedev et al.98 and Cui and Zhao99 showed that the liquid crystallinity and photo-orientation capability of covalently functionalized side-chain polymers can be modified by hydrogen bonding small molecules to the side-chain extremities. In both cases, the inscribed anisotropy was temporally relatively stable (up to remnant anisotropies of 80% or more), like in covalent azobenzene-containing polymers reported earlier.33 Contemporarily, Priimagi et al. demonstrated that the aggregation of Disperse Red 1 (DR1) molecules can be suppressed by a polymer backbone with a complementary supramolecular binding site for DR1, using both hydrogen bonding and proton transfer interactions.89 This concept was further applied to show that photoinduced birefringence and its temporal stability can be enhanced in amorphous hydrogen-bonded materials as compared to non-bonding polymer–azobenzene mixtures.90 In another highlight of supramolecular photoresponsive material design in the early years of the field, Faul, Stumpe et al. showed that the ionic self-assembly of an azobenzene small molecule (Ethyl Orange) with surfactants can create liquid-crystalline supramolecular complexes yielding temporally stable anisotropic molecular orientation, with dichroic ratios of up to 50 under illumination with linearly polarized light.100,101 To the best of our knowledge, this is still the highest photoinduced dichroic ratio reported to date. Thus, supramolecular photoresponsive materials are capable of competing with, and even surpassing, the performance of covalently functionalized materials. In this chapter, we will first concentrate on the design guidelines for ionic complexes, then for hydrogen-bonded, and finally for halogen-bonded complexes.To examine the design guidelines for ionically bonded photoresponsive materials, Bazuin and coworkers prepared the library of materials pictured in Fig. 3.82 They found that flexible molecular units are detrimental, in proportion to the degree of flexibility, to the extent and stability of the photoinduced birefringence. Thus, the most rigid structure, MO/PVP, resulted in the highest photoinduced birefringence, while both this complex and H/PVP, where the OH tail allows intermolecular H-bonding, yielded temporal stability of more than 98% of the saturated birefringence over the time period studied (Fig. 3). An additional confirmation of the benefits of such “no-spacer” structures was reached when examining the effect of the flexible spacer length between the polymer backbone and the azobenzene unit to the photoinduced birefringence (MO/PVP vs. MO/PDM), again demonstrating the detrimental role of flexible moieties on both saturated birefringence and its temporal stability in such materials.81 Accordingly, the presence of a hexyl spacer in the polymer probably contributed to the low photoinduced birefringence and its 10–20% relaxation observed for the smectic A-type LC complexes of Medvedev et al.98
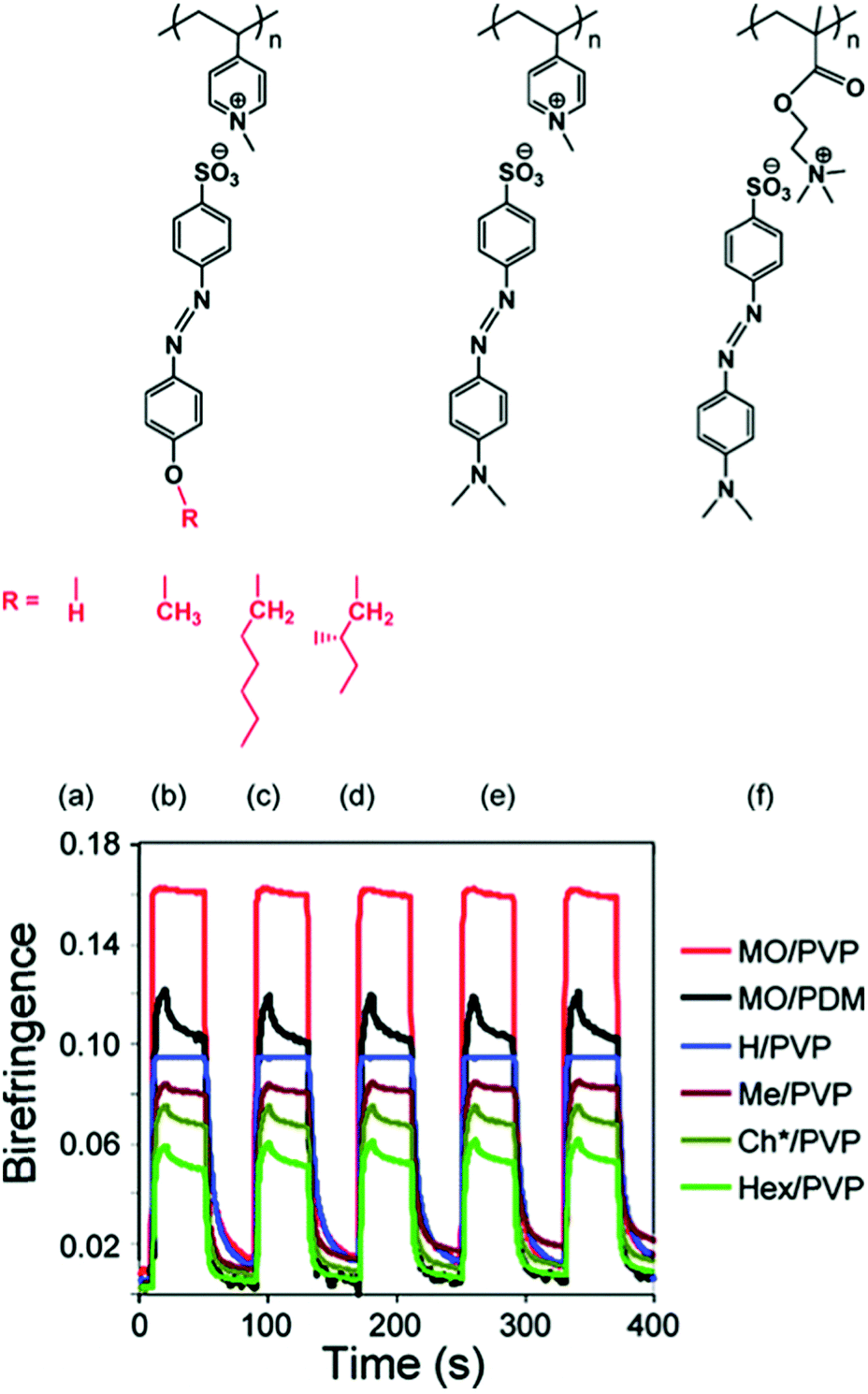 | ||
| Fig. 3 Top: Library of the ion-exchange polymer–azobenzene complexes studied, identified by the following acronyms: (a) H/PVP, (b) Me/PVP, (c) Hex/PVP, (d) Ch*/PVP, (e) MO/PVP, and (f) MO/PDM, where PVP and PDM refer to the methylated polyelectrolyte backbones. Bottom: Photoinduced birefringence cycles of (i) inscription with a linearly polarized light for 10 s, (ii) relaxation (beam off) for 30 s, (iii) erasure under circular polarization for 30 s, and (iv) beam off for 10 s in spin-coated films of the complexes. The illumination wavelength was 488 nm and intensity 320 mW cm−2. Adapted with permission.82 Copyright 2009, American Chemical Society. | ||
As another interesting strategy, demonstrated by Sanchez et al. and depicted in Fig. 4a and b, ionically bonded (proton transfer) supramolecular materials exhibiting nematic liquid crystallinity and high and stable photoinduced birefringence were obtained through acid–base titration of poly(propyleneimine) codendrimers by a COOH-capped azobenzene derivative (equimolar amounts of carboxylic acid and N-terminal amine groups).105 The best materials in terms of photoinduced anisotropy, built on a randomly hyperbranched polymer backbone of PEI-(NMe2)n, reached photoinduced birefringence of approximately 0.13 and a remarkably high order parameter of 0.67 upon irradiation with linearly polarized 488 nm light (irradiance 1 W cm−2).105 As demonstrated in Fig. 4c, the inscribed birefringence continued to increase slightly after switching off the illumination, and the inscribed birefringence was reported to be stable for several weeks.105 The same group extended the materials to a third-generation poly(propylene imine) dendrimer, which, when fully complexed with the azobenzene, CAzPA (Fig. 4a), exhibited nematic liquid crystallinity in a temperature range of 47–163 °C.106,107 The LC phase type and temperature range could be tuned by substituting CAzPA by varying amounts of a photopassive comesogen (with constant overall equimolar complexation). The dendrimer complexed with an equimolar amount of CAzPA and the photopassive comesogen showed a very broad LC temperature range, from −19 to 105 °C, and a high and stable photoinduced order parameter of 0.63.107 It was also noted that the inscribed birefringence, and thus the anisotropic molecular alignment, increased somewhat after switching off the writing beam.107
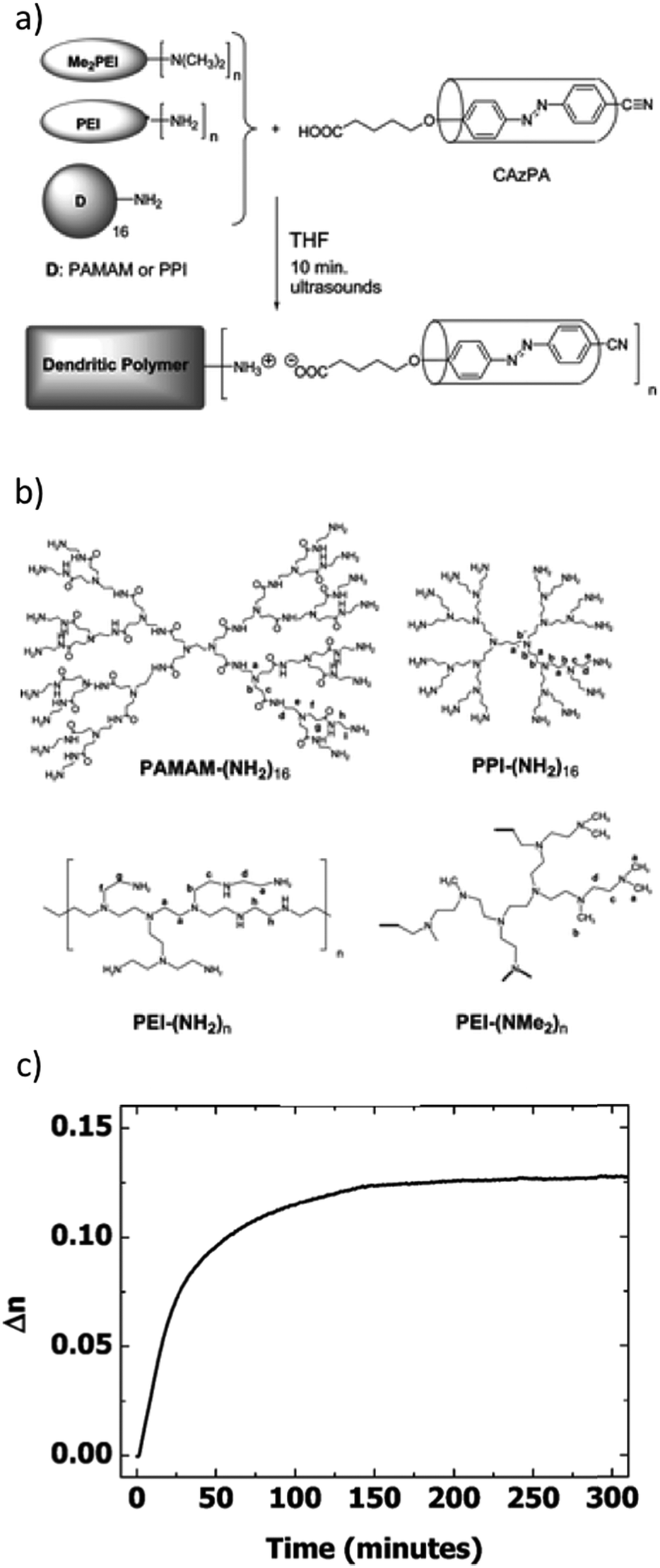 | ||
| Fig. 4 (a) Preparation method of ionic proton-transfer dendritic polymer complexes with the acid-functionalized azo derivative CAzPa, and (b) chemical structures of the dendritic polymers used. (c) Photoinduced birefringence (inscription turned on at 0 s and off at 240 s) in thin films of the complex of PEI-(NMe2)n and CAzPa. The birefringence was inscribed with linearly polarized 488 nm light at irradiance of 1000 mW cm−2 and it was observed to be stable for several weeks. Adapted with permission.105 Copyright 2008, American Chemical Society. | ||
:polymer repeat unit ratio), due to reduced aggregation.90 Later, Virkki et al. exploited the same concept in all-optical poling,110 which, unlike axial photo-orientation, leads to polar, noncentrosymmetric order of the molecules, and showed that (i) polymer–chromophore noncovalent interactions play a favorable role also in nonlinear optics, and (ii) axial and polar photo-orientation exhibit distinct concentration dependencies, the latter being more prone to chromophore–chromophore intermolecular interactions.111
The fact that DR1 tends to aggregate or phase separate at chromophore concentrations exceeding 15 mol%, when hydrogen-bonded to P4VP or poly(4-vinylphenol), served as an incitement to focus on phenolic azobenzenes and their complexes with P4VP. Hydrogen bonding between 4-nitro-4′-hydroxyazobenzene (Fig. 5a) and P4VP (denoted as P4VP(NHA)x where x indicates the phenol/pyridine molar ratio) allowed dye–polymer ratios up to equimolar to be attained without phase separation or compromising of the film quality. Direct evidence of successful complexation was obtained from IR absorption spectra (Fig. 5b) showing a systematic increase in the intensity ratio of the hydrogen-bonded pyridine band at 1009 cm−1 relative to the uncomplexed pyridine band at 993 cm−1 as a function of increasing degree of complexation. Fig. 5c illustrates that the saturated photoinduced birefringence values are systematically higher for hydrogen-bonded P4VP(NHA) complexes as compared to nonbonding PS(NHA) and P4VP(NAB) mixtures. Overall, the complexes of this study resulted in significantly higher photoinduced birefringence values (0.15 for the equimolar P4VP(NHA)1.0) compared to P4VP(DR1) complexes (0.04 for 30 wt% DR 1 in PVPh, the highest chromophore content studied).111 P4VP(NHA)1.0 also showed an impressive temporal stability of over 98%.91
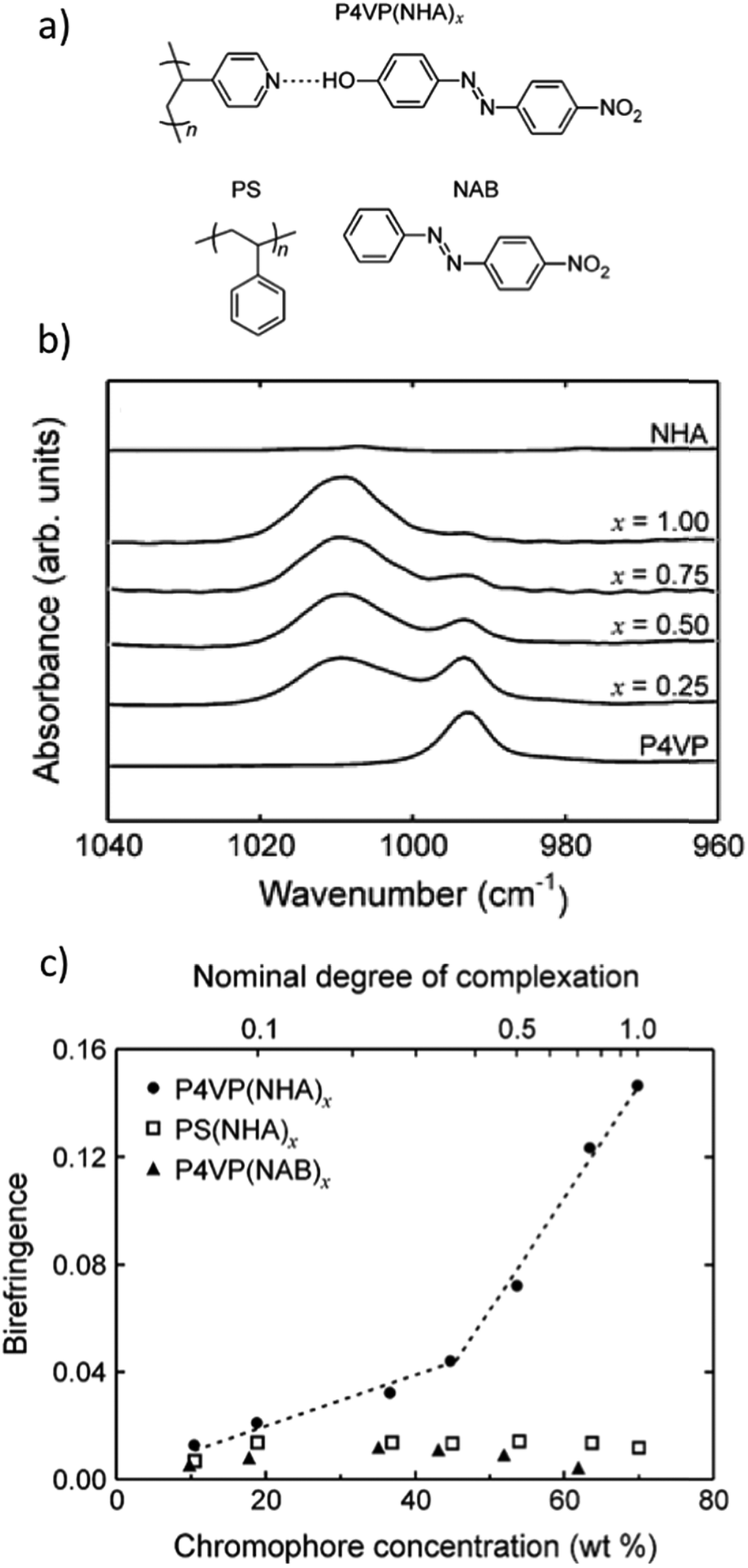 | ||
| Fig. 5 (a) Hydrogen-bonded complex between poly(4-vinylpyridine) (P4VP) and 4-nitro-4′-hydroxyazobenzene (NHA), as well as polystyrene (PS) and 4-nitroazobenzene (NAB) used for two nonbonding mixtures. (b) Infrared absorption spectra of P4VP(NHA)x complexes demonstrating the shift of the 993 cm−1 free pyridine stretching vibration band to higher wavenumbers upon hydrogen bonding and showing close to equimolar complexation for x = 1.0. (c) Saturated photoinduced birefringence (inscription done at 375 nm at 40 mW cm−2) as a function of dye concentration for the materials studied. Adapted with permission.91 Copyright 2008, American Chemical Society. | ||
Another key advantage of phenol–pyridine hydrogen bonding is that it facilitates structure–function comparisons, due to simple synthetic routes available for phenol-capped azobenzenes, allowing for building molecular libraries where one parameter at a time can be varied. Fig. 6 presents one such library, using P4VP as the polymer matrix. In all cases, the hydrogen-bonding interaction was strong enough to prevent macrophase separation of the chromophores up to equimolar ratios. This allowed assessing the effect of chromophore polarity, bulkiness, and length on photo-orientation.91,94,112–115
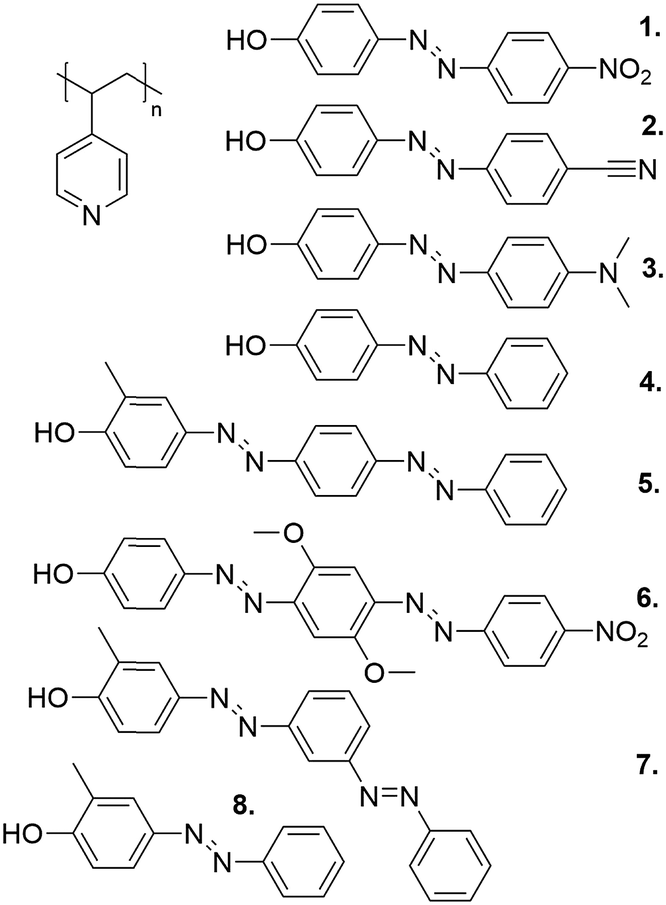 | ||
| Fig. 6 The structures of the library of phenol–pyridine hydrogen-bonded polymer–azobenzene complexes studied in the course of our work. | ||
Focusing first on molecules 1–4 (1 = NHA above), which differ only in their substitution at the tail (nonbonding) end of the azobenzene, it was shown that this change was enough to make a drastic difference in the photoresponsive behavior of the complexes. Fig. 7a shows the saturation values for photoinduced birefringence normalized by the approximate number density of the chromophores in the P4VP(2–4) complexes over a large range of complexation degrees. Such data can be used as a measure of the contribution per individual chromophore to overall birefringence.31 The most notable difference between the materials is that for P4VP(2) (the same behavior was observed also for P4VP(1),91 reported earlier) an individual chromophore contributes more to the overall birefringence at complexation degrees exceeding 0.5, whereas for P4VP(3–4), the greatest contribution per chromophore occurs at very low degrees of complexation where the chromophores presumably do not interact with one another. This can be rationalized by the fact that 1 and 2, bearing polar end groups, promote LC order in the complexes at concentrations above about 50 mol%. The onset of the appearance of an LC phase with increasing chromophore concentration can also be observed as an increased blue-shift of the UV-visible spectra of the complexes (Fig. 7b), indicative of lateral interactions between the chromophores. In contrast, molecule 3, although having a comparable dipole moment, has a bulkier tail, and molecule 4, having a somewhat lower dipole moment, result in isotropic polymer films up to equimolar complexation.94 These results indicate that intermolecular chromophore–chromophore interactions, leading to the formation of anisotropic LC domains, enable cooperative photo-orientation of the azobenzenes within the supramolecular polymeric complexes, thereby increasing the saturation values of the birefringence through combined photo-induced orientation and “dipolar dragging” of adjacent chromophores.
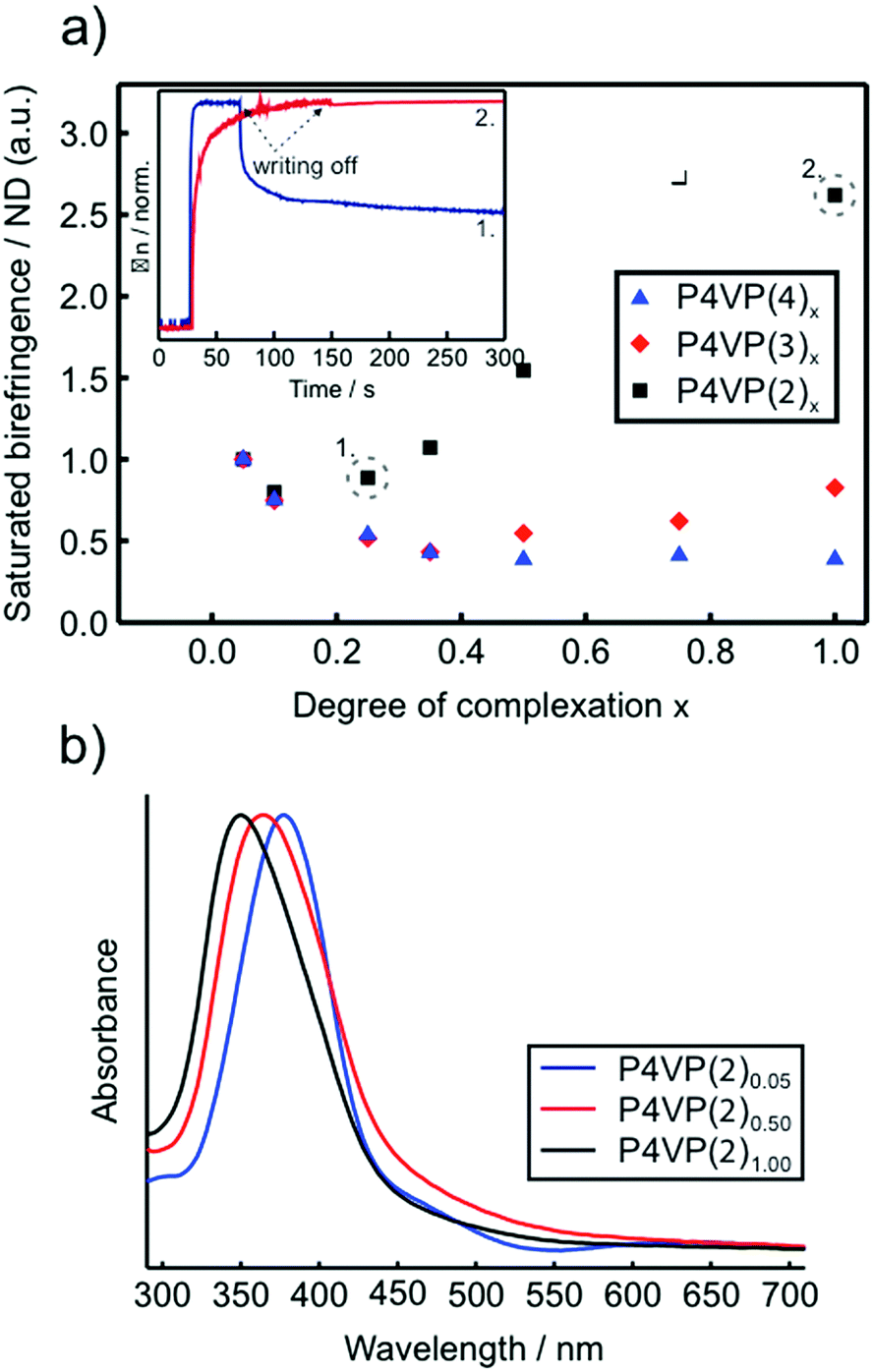 | ||
| Fig. 7 (a) Photoinduced birefringence (inscription done at 457 nm at 150 mW cm−2) normalized by the chromophore number density, i.e., the relative birefringence contribution per azo molecule, for P4VP(2–4)x. For clarity, the lowest-concentration data point (x = 0.05) is normalized to unity. Inset: Writing/relaxation curves of the photoinduced birefringence for P4VP(2)0.25 (blue curve) and P4VP(2)1.00 (red curve). (b) Normalized UV-vis absorption spectra of spin coated thin films of selected P4VP(2)x complexes. Adapted with permission.94 Copyright 2011. Royal Society of Chemistry. | ||
Similar behavior was observed by Wu and coworkers in a covalently bonded side-chain azopolymer bearing azopyridine units that were hydrogen-bonded to another azobenzene to yield supramolecular bisazobenzene complexes.116 At an equimolar degree of complexation, photoinduced birefringence of 0.1 was reported (inscription done at 442 nm with an intensity 317 mw cm−2), which increased slightly after ceasing the irradiation.116 By varying the polarization and intensity of the writing beam in a two-dimensional plane, four-dimensional optical memories with a storage density of 0.93 Gbit cm−2 (corresponding to approximately 20 times the information density of a typical DVD) were demonstrated. The stored information could be read with a polarized optical microscope. They could also be erased by heating, and subsequently rewritten.116
An infrared technique called polarization-modulation FTIR structural absorbance spectroscopy (PM-IRSAS),127 which can decouple the orientational effects originating from different chemical groups of the material system, was used to follow the molecular-level kinetics of photo-orientation induced by linearly polarized light and its subsequent thermal relaxation in the complexes of 2 and 4 with P4VP.117 This study showed that, while the saturated orientation per azo of both 2 and 4 decreases with increasing azo content, 2 orients more than 4 and only 2 also drives photo-induced orientation of the passive H-bonded pyridine groups of P4VP. Furthermore, it was concluded that 4 undergoes mainly angular hole burning resulting from angular-dependent trans–cis isomerization, whereas for 2 there is a significant additional contribution from angular redistribution. The latter, promoted by azo–azo interactions, leads to reorientation of the trans isomers of 2 and the pyridine groups to which they are H-bonded, as well as to greater orientational stability. These results explain the contrasting photoinduced birefringence results between P4VP(2) and P4VP(4) shown in Fig. 7 and illustrate how small differences in the azo molecule (the tail group here) can lead to different photo-orientation mechanisms.
Complexes of P4VP(5–8) were found to be isotropic throughout the complexation range studied, probably due to rotating side groups and/or nonlinear shape, both hindering molecular packing. Even though isotropic, photo-orientation was observed in all of these materials, and when comparing P4VP(5) to P4VP(8), it was shown that the larger aspect ratio of 5 gave rise to a 4-fold increase in birefringence when using the same azobenzene concentration.115 Also, two conclusions of practical importance for the selection of inscription wavelength were drawn. First, the electronic structure of 6 greatly broadened the absorption spectrum, making it possible to inscribe birefringence over a wide range of wavelengths, from 375 nm to 633 nm.112 Second, the writing wavelength comparison for P4VP(7) and P4VP(8) showed that, in order to reach significant birefringence levels, it is important to use the wavelength that drives the photoisomerization reaction in both isomerization directions (trans-to-cis and cis-to-trans), even when that wavelength (488 nm) lies far from the absorption maximum (356 nm for P4VP(7) and 354 nm for P4VP(8)).115 In general, the ability to record birefringence at wavelengths barely absorbed by the material may be beneficial for applications such as optical holography,118,119 where low penetration depth of conventional azobenzene-containing polymers may become a limiting factor.
One pertinent question provoked by these studies is the role of the strength of the hydrogen-bonding interaction. With this in mind, we attempted to extend the library of azobenzenes to a COOH-capped and an SH-capped dye acting as a stronger and a weaker hydrogen-bond donor, respectively. However, experimental problems were encountered due to the tendency of the COOH-capped azobenzenes to self-associate and SH-capped ones to dimerize through S–S bonds, thus leading to crystallization and phase separation in the polymer matrix. A similar tendency for COOH-functionalized azobenzenes in P4VP was observed by del Barrio et al., who showed that phase separation occurs for degrees of complexation greater than about 0.50 for a carboxy-capped cyanoazobenzene containing a flexible alkyl spacer.120
To circumvent the inherent dimerization tendency of carboxylic acid-functionalized azobenzenes at higher degrees of complexation, a dendritic molecule combining three azobenzene moieties, dAZO, was synthesized and hydrogen-bonded to P4VP, as shown in Fig. 8.121 This allows a high number of active azobenzene units for a lower number of hydrogen-bonding units. Another strategy to inhibit COOH dimerization is to modify the backbone, allowing it to accept multiple hydrogen bonds from one tecton by polymerizing a methacrylic monomer containing a 2,6-diacylaminopyridine unit, thus increasing the total amount of dye loading by suppressing the phase separation of tectonic units at degrees of complexation exceeding 0.50.122 The reached 1:1 (tecton:polymer repeat unit) complex, which is liquid crystalline, yielded essentially stable photoinduced anisotropy.122
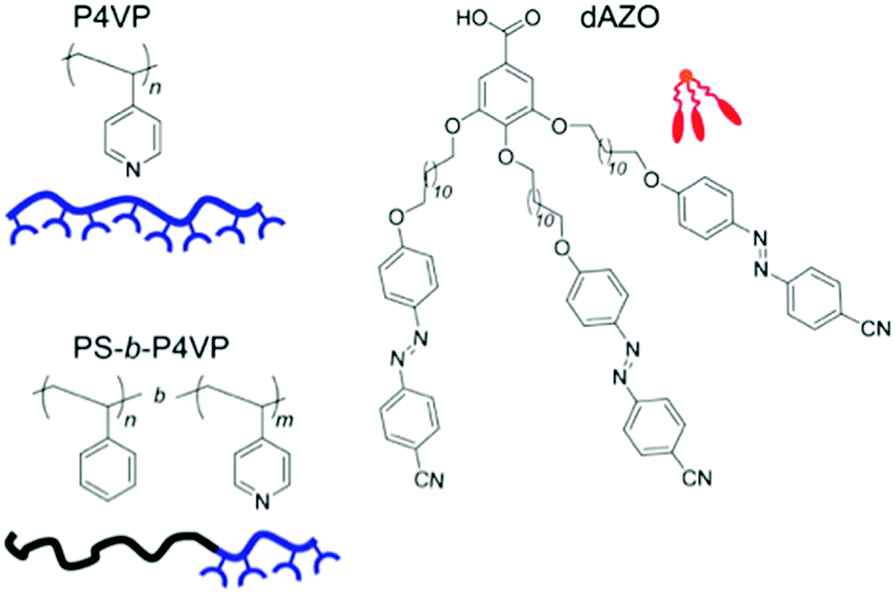 | ||
| Fig. 8 Azodendron dAZO composed of a benzoic acid derivative with 3 alkoxy-cyano-azobenzene substituents, and structures and schematic representations of P4VP homopolymer and PS-b-P4VP block copolymer. Adapted with permission.121 Copyright 2014, American Chemical Society. | ||
To further compare halogen bonding with hydrogen bonding, we employed PM-IRSAS to show that the passive pyridine moiety in P4VP can rotate in concert with the photo-orienting azobenzene.84 Even though the interaction strength of halogen bonding is less than that of hydrogen bonding, it gave rise to more pronounced pyridine orientation upon illumination. This was attributed to the much higher orientation of the halogen-functionalized azo unit, which in turn was related to the higher Tg of the halogen-bonded complex.84 The advantage of the more directional nature of halogen bonding has also been pointed out in nonlinear optics. A simple mutation of the halogen from iodine to bromine in halogen-bonded azobenzene–P4VP complexes more than doubled second-harmonic generation upon all-optical poling of the materials.128
The choice of bonding type depends also on what the user is aiming for. Although the highest photoinduced anisotropy thus far has been observed in complexes of small molecules,100,101 the polymer materials reviewed here have the advantage of easy processability and good film-forming capacity. Generally, increasing the number of photoactive azobenzene units will increase the saturation value of the photoinduced birefringence (provided that no phase-separation of the chromophores occurs); however, for some applications, lower azobenzene content and thus lower absorption (or thicker films enabled by the lower absorption) may be preferred. Halogen bonding, as the newcomer in this field, is very promising, given its highest directionality of the three bonding types under consideration.
3.2. Photoinduced surface patterning
The first demonstrations that hydrogen bonds97,108,109 and ionic bonds93,102,129 can be used to create materials that undergo efficient photoinduced surface patterning were published in 2007 and early 2008. Zettsu et al. showed unambiguously, by comparison with nonbonded or poorly bonded mixtures, that the supramolecular link between the photoactive units and the passive polymer matrix is essential for the formation of surface-relief structures in liquid crystalline hydrogen-bonded polymer–azobenzene complexes.97 It thus functions similarly to covalent bonds, considering that, for example, Lagugné Labarthet et al. had pointed out that covalently functionalized polymers result in surface-relief gratings that are superior in terms of modulation depth as compared to those in dye-doped polymers.130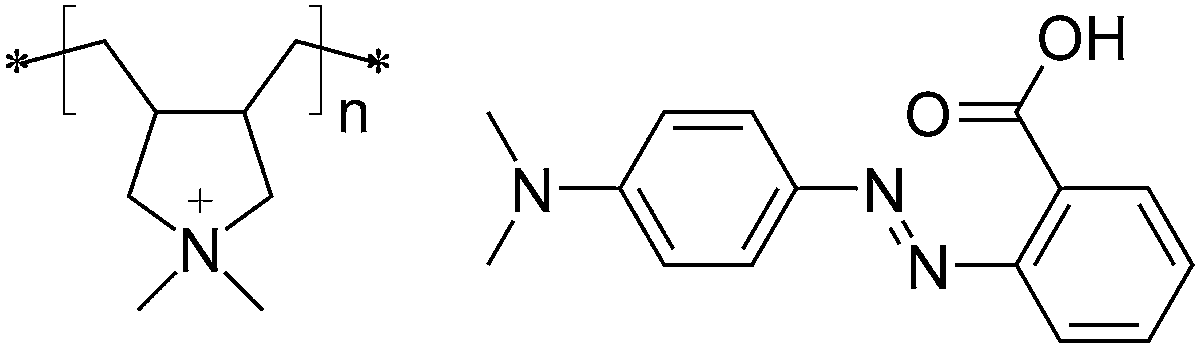 | ||
| Fig. 9 Chemical structures of the constituents, poly(diallyldimethylammonium chloride) (PDADMAC) and methyl red (MR), of one of the ionically bonded polymer–azobenzene complex studied in ref. 129. | ||
The characteristics of efficient SRG formation in the timescale of minutes (at lower irradiance) were similarly observed for the MO/P4VP complex (Fig. 3), for which gratings of 360 nm modulation depth, exceeding the original film thickness of 305 nm, could be written within 5 min with the relatively low irradiance of 65 mW cm−2.82,93 The high photoinduced surface patterning efficiency was attributed to the strong ionic interactions, the molecular rigidity, the very high Tg, and the LC nature of the complex. When comparing the MO/P4VP complex with the other ionically bonded materials presented in Fig. 3, the same correlation as for photo-orientation was found: the more rigid the complex, the more efficient the SRG formation. From this point of view, the complex in Fig. 9 is also a very rigid system, leading to very efficient SRG formation. It should be emphasized that the polymer molecular weight in both types of ionic complexes was high (200000 for P4VP82 in Fig. 3 and ca. 500000 for PDADMAC in Fig. 9129).
In addition to linear polyelectrolyte backbones, dendritic polyelectrolyte backbones have been used as building blocks for ionic dendrimer–azobenzene complexes.134,135 As opposed to linear polymer chains, dendrimer structures allow increasing the molecular weight and azobenzene content in the material while retaining the globular shape of the molecules.136 As indicated in Fig. 10, up to very large globular supramolecular complexes of generation 10 could be prepared and made to move in response to light.134 These molecules can be considered as model systems for large biomolecules such as native proteins, which, when complexed with azobenzenes, could also undergo photoinduced mass transport.134
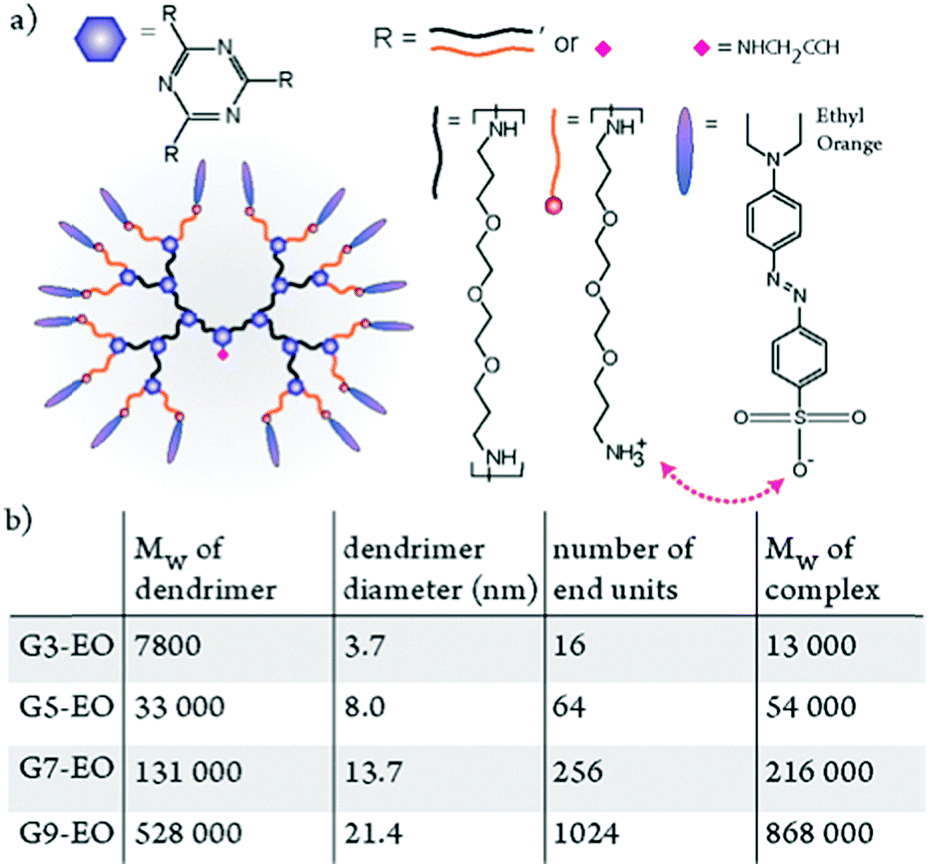 | ||
| Fig. 10 (a) Chemical structure of an ionically bonded complex of ethyl orange (EO) and a third-generation dendrimer. (b) The nominal molecular weight and experimentally determined diameters of the dendrimers, the number of binding sites in each dendrimer, and the molecular weight of nominally stoichiometric dendrimer–EO complexes. Adapted with permission.134 Copyright 2014, American Chemical Society. | ||
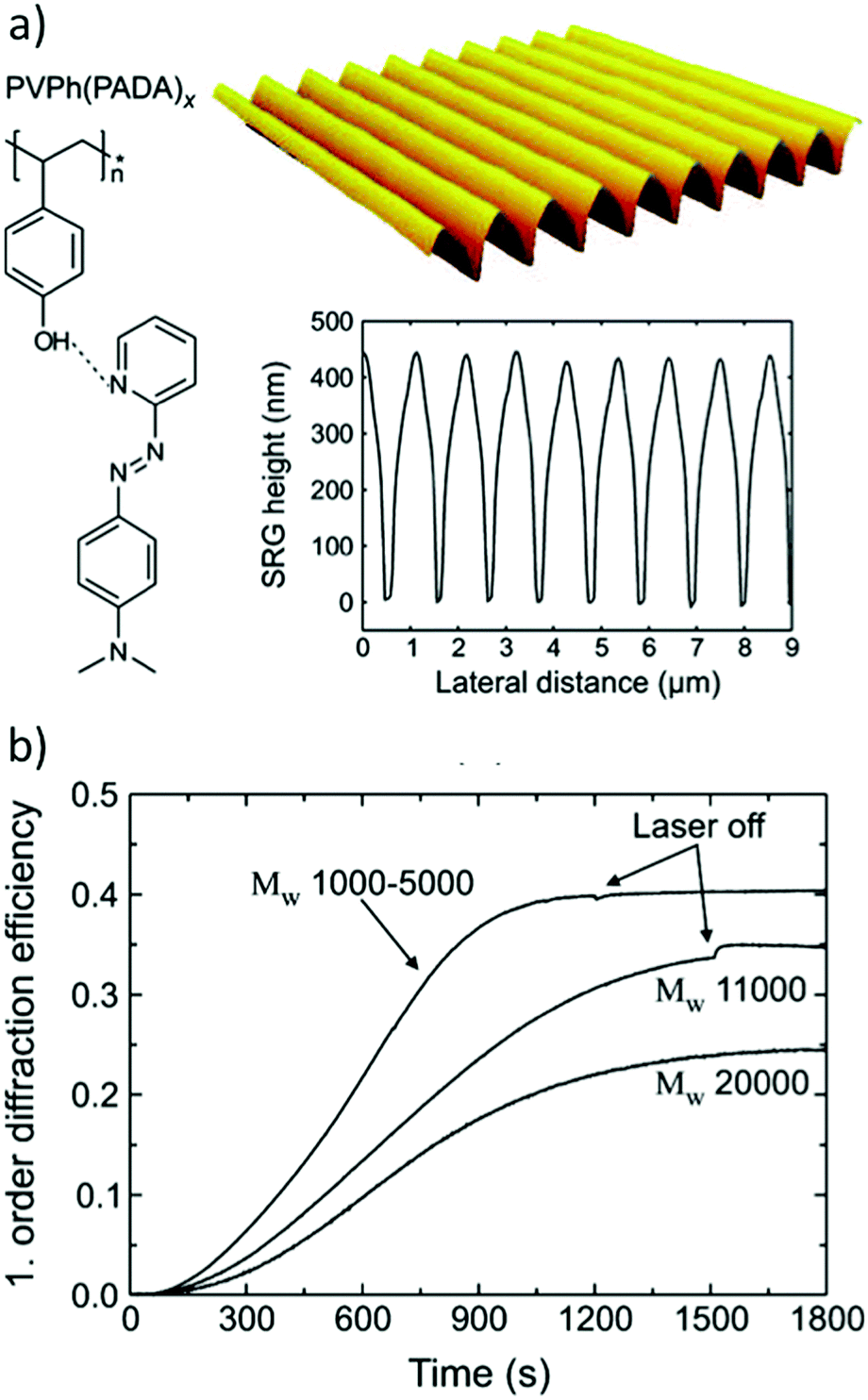 | ||
| Fig. 11 (a) Molecular structure of a hydrogen-bonded poly(vinylphenol)–azobenzene complex, an example of an inscribed SRG and a cross-section of an inscribed SRG determined by AFM. (b) The first-order diffraction efficiency during SRG inscription for three different molecular weights of the polymer backbone. Adapted with permission.137 Copyright 2009, American Chemical Society. | ||
Regarding the library of hydrogen-bonded materials presented in Fig. 6, it was found that the lateral intermolecular interactions leading to formation of LC domains with enhanced photo-orientation, counteracted the formation of photoinduced surface patterns. In particular, of the monoazo compounds 2–4, the amorphous P4VP(3) complexes were found to be superior in SRG inscription efficiency as compared to P4VP(2) or P4VP(4).94 When comparing the bisazobenzene complex P4VP(7) in Fig. 6 with the analogous monoazo complex, P4VP(8), it was found that SRG inscription in the former is more efficient than in the latter. This was attributed to the fact that photoisomerization of a bulkier molecule requires a larger sweep volume and thus induces more motion into the material.115 Moreover, similarly to photo-orientation, the wide absorption spectrum of P4VP(5) made it possible to inscribe SRGs over a very broad spectral range, from 405 nm to 633 nm.112
For some subsequent studies on hydrogen-bonded materials, P4VP(3) was selected as the standard. This is because it remains amorphous, with the chromophore well dispersed into the polymer, up to equimolar complexation. One recent study concentrated in finding the lower limit of the amount of azobenzene needed to create SRG patterns. It was found that as little as 1 mol% (2.2 wt%) of 3 was enough to result in a grating with a modulation depth of 170 nm.113 This surprising result indicates that even if 9 out of 10 polymer chains (99 out of 100 repeat units) are statistically free of azobenzene, macroscopic mass transport nevertheless occurs. This provokes the question as to whether the hydrogen bonds are “photodynamic”, hopping between different polymer chains, while still strong enough to drive mass transport, an issue that still needs to be clarified.
The same material system, P4VP(3), was used to confirm that the SRG formation efficiency depends inversely on the chain length of polymers, and thus on Tg, both with conventional interference patterns of circularly polarized light138 and with a nonconventional s–s interference pattern, which previously had resulted in very poor SRGs.139 These gratings could also be erased by illuminating them with uniform light to induce inverse mass transport. Using light as an erasing tool instead of the more commonly used heating method has the advantage that light has a much higher spatial and temporal resolution, thus allowing more complex surface patterns to be prepared, as demonstrated in Fig. 12.138 In addition, this material has been used in recent explorations of photo-induced willow structures under uniform single beam illumination but with substantially higher incident irradiance as compared to SRG inscription.140,141
 | ||
| Fig. 12 Scanning electron microscopy (SEM) images of surface patterns with spatially varying grating vector directions, created by subsequent optical inscription and erasure steps. Reprinted with permission.138 Copyright 2015, Royal Society of Chemistry. | ||
Sobolewska and coworkers explored a library of different hydrogen-bonded polyimides and their capacity for photoinduced surface patterning.142,143 While acknowledging that more work is required to establish conclusive structure–property design guidelines for the polyimide-based complexes, they clearly showed that the chemical structure of the backbone and whether it contains OH or COOH groups (the latter being subject to intra- and intermolecular hydrogen bonds between polymer repeat units) are among the determining factors for photoinduced surface patterning efficiency. This highlights the fact that photoinduced surface patterning requires the motion of the whole material system, and thus the interactions between polymer chains may be as important, or in some cases even more important, than the interactions between the polymer host and azobenzene guest.
The effect of hydrogen bonding on SRG formation efficiency has been compared recently with that of proton-transfer ionic bonding and of pure ionic bonding obtained by ion exchange, using the analogous complexes shown in Fig. 13.83 The Tg of the starting polymer is low (20 °C) but increases with degree of complexation and is very high for the purely ionic complex (180 °C). As shown in Fig. 13, the proton transfer complex, despite some crystallization of the azobenzene molecules, led to greater SRG formation efficiency than the H-bonded complex, whereas, most impressively, the ion-exchange complex led to much greater SRG formation efficiency. This result suggests that ionically bonded and H-bonded complexes may have an inverse dependence of SRG efficiency on molecular weight and the Tg and that the role of these parameters in SRG formation is not yet fully understood.
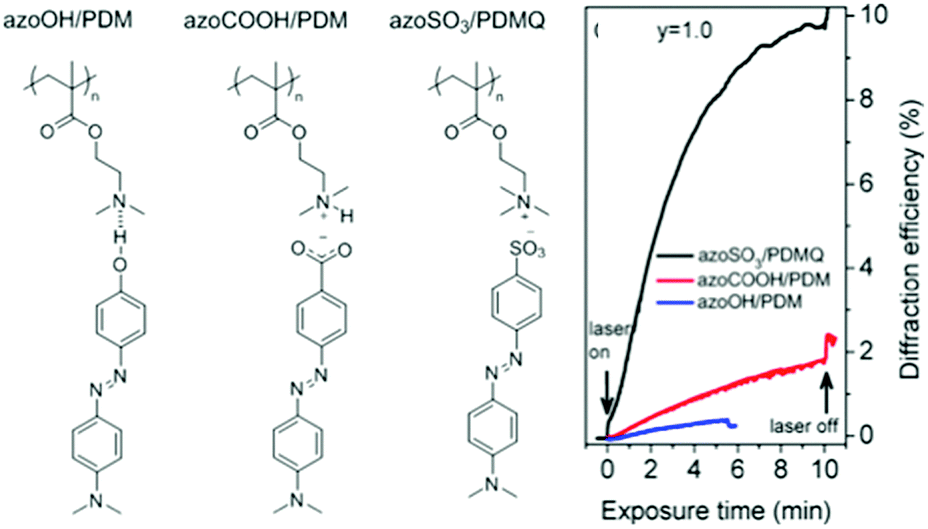 | ||
| Fig. 13 Chemical structures of analogous hydrogen-bonded, proton transfer and ion exchange complexes, as well as the first-order diffraction efficiency upon SRG inscription. Adapted with permission.83 Copyright 2016, American Chemical Society. | ||
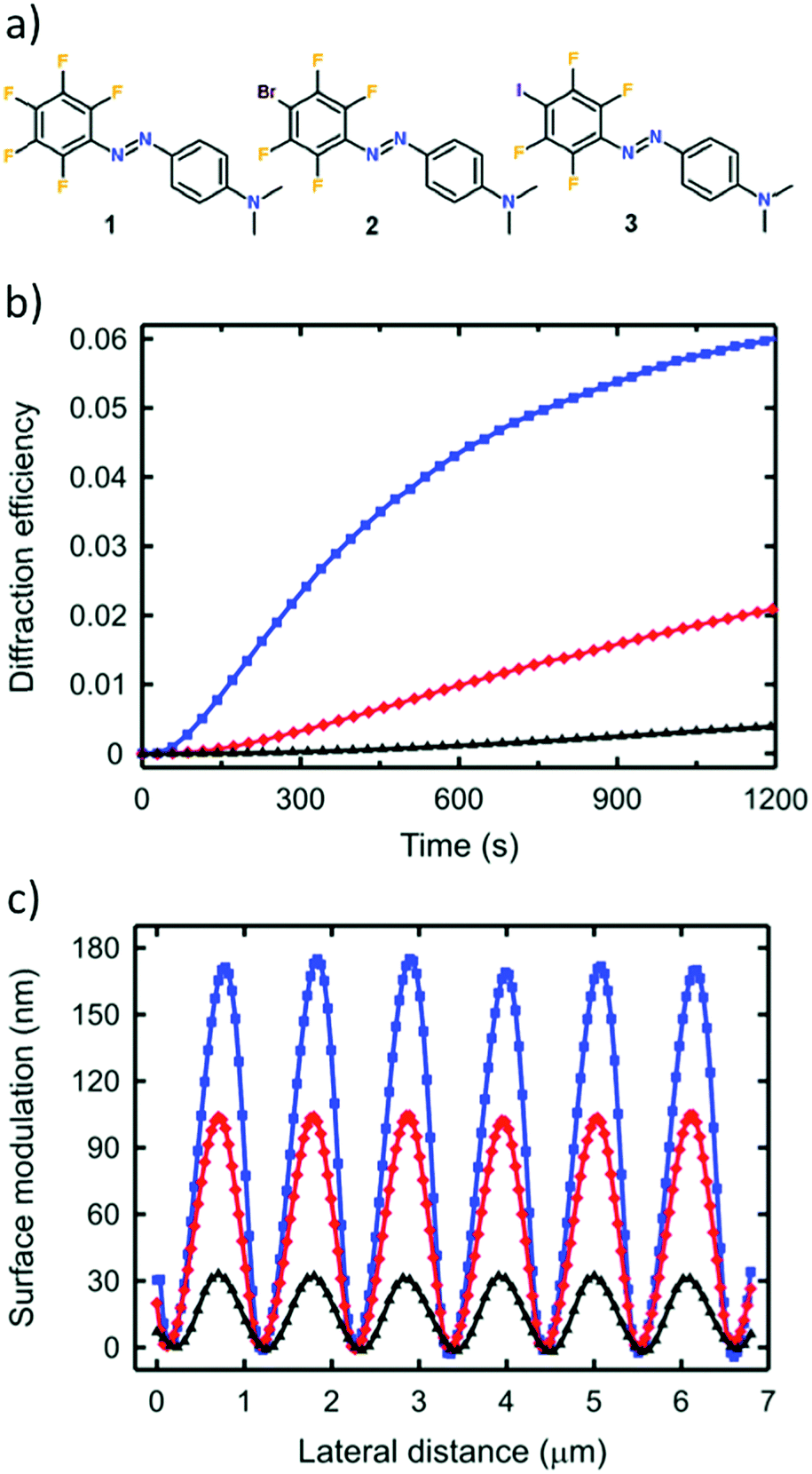 | ||
| Fig. 14 (a) Chemical structures of the halogen-substituted azobenzenes used. (b) First-order diffraction efficiency and (c) resulting sinusoidal modulation depth of surface-relief gratings on films of halogen-bonded P4VP–azobenzene complexes, for which the computed halogen-bonding interaction strength between ethylpyridine and the chromophores are −5.1 kcal mol−1 (chromophore 3, blue), −3.5 kcal mol−1 (chromophore 2, red) and 0 kcal mol−1 (chromophore 1, black). Adapted with permission.145 Copyright 2015, Royal Society of Chemistry. | ||
Like for the inscription of photoinduced anisotropy, the choice of material for surface structuring depends on the needs of the application in question. If thermal and chemical stability is sought for, or if high SRG formation efficiency coupled to high photo-induced anisotropy is needed, ionic complexes are the materials of choice. If the azobenzenes are to be selectively removed after, for instance, an SRG is formed, weaker hydrogen- and halogen-bonding are better adapted to such removal. Also, to our knowledge, photoinduced erasure of SRG patterns that works well for hydrogen-bonded polymer–azobenzene complexes of relatively low azo content,113 has not yet been systematically studied in ionic or halogen-bonded complexes.
3.3. Photocontrol of micro- and nanostructures through supramolecular strategy
Supramolecular control has also been used for block copolymers (BCPs), which generally self-organize into domains with characteristic length scales of 10–100 nm due to block immiscibility.13 If one block bears units that are suitable for supramolecular bonding, the small molecules comprising a complementary functionality will selectively reside in that block, thus changing the relative block volume ratio and often altering the BCP morphology. Pioneering studies along these lines were done by Ikkala, ten Brinke and coworkers, using, for example, phenol–pyridine hydrogen bonding between functionalized alkyl chains such as pentadecylphenol (PDP) and PS-b-P4VP (PS:polystyrene).147 However, none of the BCP complexes they prepared involved azobenzene guests. On the other hand, Xu and co-workers,148 Stamm and coworkers,149–151 and, recently Gröschel and coworkers,152 have used azobenzene-containing hydrogen-bonded side chains in investigations involving thin-film nanopatterning, nanoparticle assembly, and templating with supramolecular BCPs, but they did not explore the potential photoresponsive properties of these complexes.Photoinduced control over the phase structure of BCP materials combined with supramolecularly bonded azobenzene molecules is a highly interesting phenomenon, which has only recently begun to be explored. One of the first studies was reported by del Barrio et al., who showed that, when photoinduced anisotropy is inscribed in self-assembled BCP structures composed of PS-b-P4VP and an azobenzene hydrogen-bonded to the minority P4VP block, the extent of photo-orientation is dependent on the block copolymer morphology, and lamellar structure resulted in higher photo-orientation than spherical structure.120 To further highlight the effect of self-assembly on anisotropy, both the photoinduced birefringence and order parameter in self-assembled structures were measured to be smaller than in an analogous amorphous P4VP–azobenzene complex, thus suggesting that a rigid polymer environment influences the efficiency of photoinduced processes.120
Inscribing SRGs into BCPs creates its own challenges and opportunities, since the azobenzene-containing phase is not necessarily continuous. In 2006, the photoinduced alignment of a self-assembled cylindrical morphology upon SRG inscription in a covalently-synthesized azobenzene-containing diblock copolymers was observed,153 and the fundamental understanding of these alignment processes has recently been reviewed by Seki.154 Applying UV intensity interference patterns to diblock copolymer brushes covalently attached to flat solid substrates and ionically complexed with azobenzene, it was found that the local variation in azobenzene photoisomerization leads to the formation of SRG patterns accompanied by optomechanical scission.155 One interesting SRG-forming azobenzene-containing BCP is a polyelectrolyte block copolymer composed of two different poly(ferrocenylsilane) blocks, one of which contains azobenzene.69 This material exhibited a cylindrical microphase-separated structure, with the azobenzene-containing block forming the continuous phase. The SRG modulation depth was shown to be enhanced by solvent annealing, and furthermore, the structure could be swollen by oxidizing the ferrocene units, resulting in a significant increase in the SRG modulation depth. Finally, the authors showed that the organic parts of these polymers can be removed via plasma treatment, leaving behind an etch-resistant ceramic grating.69
Recently, Vapaavuori et al. demonstrated that the BCP morphology of a thin film containing a block-selective hydrogen-bonding azobenzene can be manipulated with light.156 As explanatory background, some of the authors had previously shown that when the films are prepared using the dip-coating technique (i.e. where a substrate is pulled out of the polymer solution at a given rate), the hydrogen-bonding small molecule content in the film is controlled by solution azo/polymer repeat unit ratio, dip-coating withdrawal speed, and the solvent used.157,158 The latter two are especially critical at very slow dip-coating speeds in the so-called capillarity region, and can cause film morphology changes as a function of dip-coating rate. Vapaavuori et al. took advantage of this dependence by comparing the film patterns obtained by dip-coating, under illumination and without illumination, from a THF solution of PS-b-P4VP and 4-butyl-4′-hydroxyazophenol (BHAB) in a 0.25 molar ratio relative to the pyridine repeat units.156 Normally, dip-coating of such supramolecular solutions without illumination at slow dip-coating rates gives rise to films whose small-molecule content is much lower than in solution but increases with increasing dip-coating rate, as observed also with BHAB. This led to a change in film morphology from spherical to cylindrical at a dip-coating rate of around 1.0 mm min−1 for the system studied. When this same system is dip-coated while being simultaneously illuminated by a 365 nm LED, the cylindrical morphology was obtained already at 0.5 mm min−1. A comparison of the surface morphologies of the films obtained at this dip-coating rate, showing dots (indicative of spheres) and stripes (indicative of horizontal cylinders) for dip-coating with the light off and on, respectively, is shown in Fig. 15. It was also noted that the BHAB content in the dip-coated film is increased by dip-coating under illumination compared to dip-coating in the dark. This increase, along with the greater volume associated with the cis isomer, rationalizes the observed photoinduced morphology change.
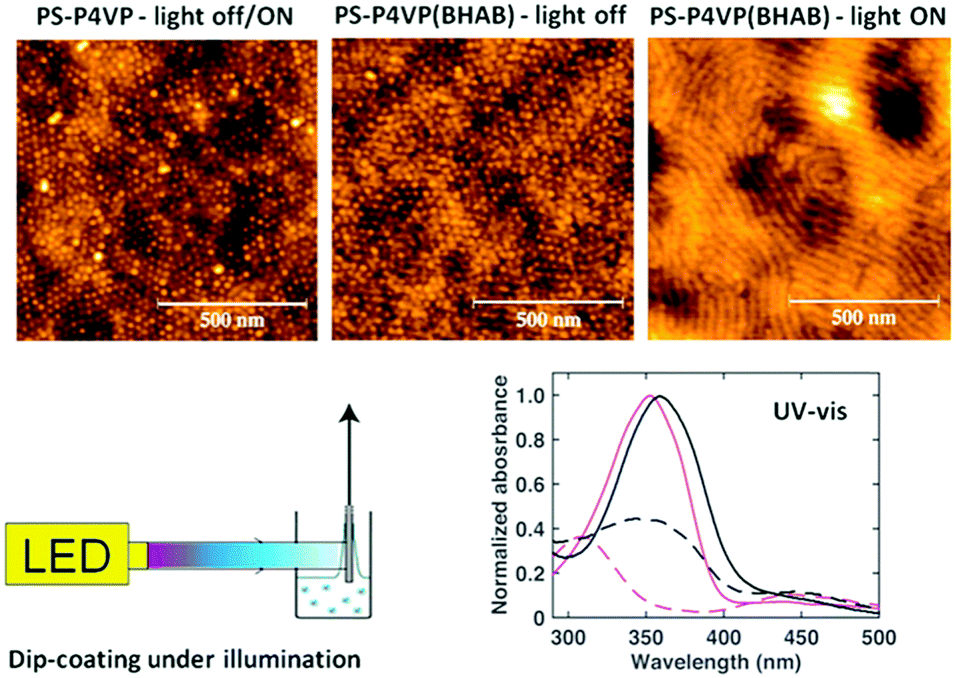 | ||
| Fig. 15 Topographical AFM images (1 × 1 μm2) of thin diblock copolymer films of PS–P4VP dip-coated in the dark, PS–P4VP(BHAB), dip-coated in the dark, and PS–P4VP(BHAB) dip-coated under 365 nm LED irradiation, all at a withdrawal rate of 0.5 mm min−1 from THF solutions. Schematic diagram of the dip-coater setup and UV-visible spectra of a PS–P4VP(BHAB) solution (magenta) and spin-coated thin film (black), before (solid lines) and directly after (dashed lines) irradiation. Adapted with permission.156 Copyright 2015, American Chemical Society. | ||
These results show that light can be used to command the nanostructured morphology of dip-coated thin films of supramolecular BCPs. An additional advantage of the supramolecular strategy was also demonstrated; namely, that the BHAB molecules can be removed from the films after dip-coating by selective solvation without changing the morphology. This manipulation renders the films optically transparent and is not available for conventional covalently bonded azobenzene side-chain polymers.
Another way of combining supramolecular material design and top-down photocontrol of the 3D structures was demonstrated by Yu et al., who succeeded in preparing light-deformable liquid crystalline microparticles via a supramolecular strategy, notably by employing an azopyridine-containing polymer as a hydrogen-bonding acceptor and a homologous series of dicarboxylic acids as hydrogen-bond donors that also act as supramolecular crosslinkers.159 As illustrated in Fig. 16, application of 360 nm UV-light at a temperature of Tg + 10 °C results in both a photoinduced LC phase transition and deformation of the microparticle shape. Later, the Yu group demonstrated that an analogous material design combined with slow evaporation from a mixture of good and poor solvents produces walnut-like wrinkled photoresponsive microparticles.160 In addition, they showed that complexes of the same azopyridine-containing polymer and poly(acrylic acid) results in the formation of ultrathin Janus films exhibiting wetting properties that depend on the film side.161
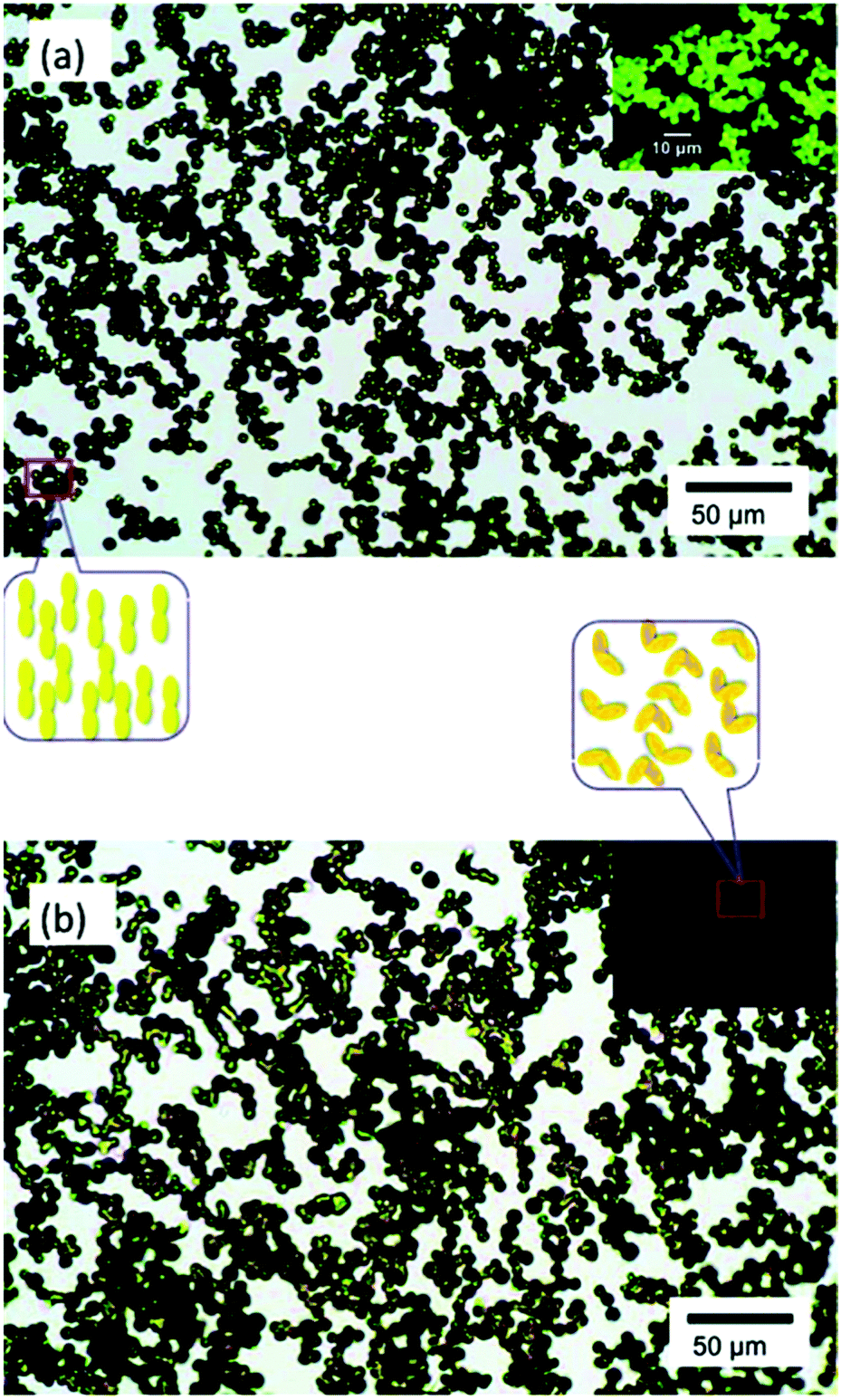 | ||
| Fig. 16 Optical micrographs of supramolecular microparticles before (a) and after (b) illumination at 360 nm. The inset illustrates through polarized optical microscopy that a photoinduced liquid crystalline–isotropic phase transition occurs simultaneously with shape deformation of the microparticles. Reprinted with permission.159 Copyright 2011, American Chemical Society. | ||
Block copolymers in solution bring an additional twist to self-assembly phenomena, since the interactions with the solvent molecules may allow formation of micellar structures or gels. By synthesizing copolymers of thermoresponsive pNIPAM and carboxylic-acid-functionalized azobenzene-containing monomers, the Yu group were able to discover a region, in which fluorescence from aggregated azobenzene in the micellar structures can be enhanced by photoisomerization with UV-light.88 Moreover, the fluorescence of the aggregates responds to changes in temperature and pH, thus showing how clever molecular design and interplay with solvent can lead to truly multiresponsive structures. Additionally, by applying the homopolymer of carboxylic acid functionalized azopolymer in DMSO, UV-irradiation was used to trigger a gel–sol phase transition.162
Finally, hierarchical structures involving block copolymer self-assembly and laser ablation were demonstrated, as depicted in Fig. 17b.163 By employing solvent annealing of PS–P(2/4)VP containing hydrogen-bonded azobenzene molecules, as shown in Fig. 17a, vertically oriented cylindrical structures were obtained. When these were then exposed to a single laser shot interference pattern with a 35 ns laser pulse, a larger length-scale cylindrical structure was created. The smaller length scale of the BCP self-assembly could be tuned by changing the chemical composition, and the larger length scale by changing the polarization of the interference beams. Unlike the previous example, this patterning method is not based on the photoresponse of the azobenzene molecules, and is thus not limited to azobenzene-containing materials.
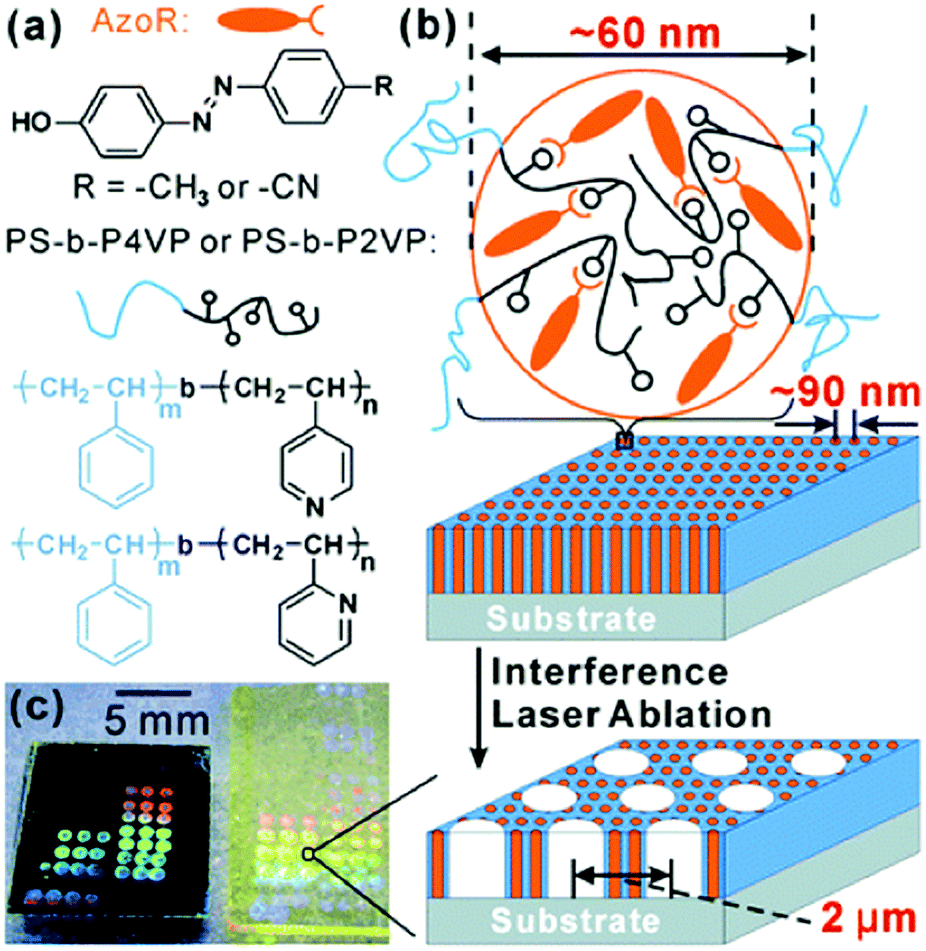 | ||
| Fig. 17 (a) Chemical structures of the employed hydrogen-bonded block copolymer complexes. (b) Schematic model of hierarchical structure formation by self-assembly of BCP cylinders (smaller scale cylinders) and laser ablation (larger scale cylinders). (c) Colors resulting from hierarchical photonic structures of these samples fabricated on a silicon wafer (left) and on a glass slide (right). Reprinted with permission.163 Copyright 2012, Royal Society of Chemistry. | ||
4. Concluding remarks and outlook
In this review, we have discussed the developments in the field of supramolecular photoresponsive polymer materials during the last decade. During this time period, the most commonly used noncovalent interactions to efficiently integrate photo-responsive small azobenzene molecules into polymers or other large amorphous macromolecules have been ionic, hydrogen and halogen bonding. In addition to strength, these three supramolecular interactions differ in nature, which is most clearly viewed in their different methods of formation and directionalities. Ionic interactions have no preferred directionality, whereas halogen bonding is highly directional due to the narrow confinement of the electropositive bond-donating area on the polarizable halogen atom.From the point of view of preparation, hydrogen- and halogen-bonded materials, as well as ionically bonded ones involving proton transfer, are more versatile, requiring simple mixing of the components in a suitable solvent followed by solvent removal. In contrast, the preparation of ionically bonded materials obtained by ion exchange is often a multi-step procedure, sometimes requiring prior polymer modification (such as quaternization) and generally requiring the elimination of the original counterions to drive the desired ionic bond. Ionic interactions also typically reduce the choice of appropriate solvents compared to hydrogen and halogen bonding. This also means that establishing a series with varying degrees of complexation for fundamental structure–property studies is more straightforward with hydrogen- and halogen-bonded materials. Additionally, many device applications demonstrated to date in covalent systems that benefit from photoresponsive azopolymers, such as photonic components,67,164 organic lasers165,166 and conductivity switching,167–169 could benefit from employing the supramolecular materials for optimizing, e.g., the azobenzene content for the described application.
Ionic bonding, on the other hand, is generally more stable thermally and temporally, whereas hydrogen- and halogen-bonded small molecules are more susceptible to partial loss at higher temperatures or under vacuum drying. The latter can also be easily removed from the final material if desired by selective solvation. The various advantages and limitations, as well as the most commonly used characterization methods for validating the formation of the supramolecular complexes and some specific properties, are collected in Table 1. We also emphasize that the supramolecular strategy allows the mixing of different chromophores, including more than one type of supramolecular bond, into the same complex, in case a larger spectral response or combinations of different functions or properties is sought.
| Interaction type | Common characterization methods | Advantages | Limitations |
|---|---|---|---|
| Ionic (ion exchange) | Successful elimination of counter ions by EDS. Verification of stoichiometry by elemental analysis, NMR. | High photoinduced anisotropy and efficient SRG formation achieved in same material. Phase separation not usually an issue up to stoichiometric chromophore content. Some materials are water-soluble. | Must eliminate original counterions, adding to the preparation steps. Tend to be humidity-sensitive. |
| Changes in material system by DSC, POM, XRD, UV-vis. | |||
| Hydrogen bonding | Easily observed by FTIR. Verification of stoichiometry by elemental analysis, NMR. | Easy preparation (simple mixing of the constituents at desired ratio). Very low chromophore content easy to achieve and surprisingly efficient in SRG formation. Easy to eliminate the azo molecules by selective solvation. | Molecular aggregation/phase separation can be an issue at higher chromophore content. Azobenzene content can be reduced by excessive heating/vacuum drying. |
| Changes in material system by DSC, POM, XRD, UV-vis. | |||
| Halogen bonding | Easily observed by FTIR and XPS. Verification of stoichiometry by elemental analysis, NMR. | Easy preparation (simple mixing of the constituents at desired ratio). Allows addressing the effect of interaction strength by changing the halogen substitution. | Molecular aggregation/phase separation can be an issue at higher chromophore content. Azobenzene content can be reduced by excessive heating/vacuum drying. |
| Changes in material system by DSC, POM, XRD, UV-vis. |
Even though the picture of structure–property–function relations for efficient supramolecular azobenzene-containing polymers is still far from complete, progress has been made in the last decade and a number of guidelines can be highlighted. In the core of these guidelines is the choice of type and strength of the noncovalent bond (or bond mixture) for optimizing the desired optical and other properties of the material system.
One guideline, highlighted in a study of ionically bonded complexes, is that rigidity (i.e. lack of flexible molecular moieties in complexes) is an important characteristic for improved optical response. Although this factor has not been directly compared in hydrogen- and halogen-bonded complexes, we note that majority of the structures of these types reported in the literature are relatively rigid “no-spacer” structures. To further assess this structure–property relation, flexible moieties should ideally be incorporated into the host material, since adding long alkyl chains to azobenzene will drastically alter its photochemical characteristics and thus not serve for the intended comparison. The success of no-spacer structures has been enabled by supramolecular design, since the preparation of such rigid structures through covalent polymer synthesis would be more complicated.
As another guideline, direct comparisons between different noncovalent interactions suggest that the SRG formation efficiency of ionically-bonded complexes surpasses that of hydrogen-bonded complexes, and halogen-bonded complexes surpasses the hydrogen-bonded ones despite the latter being a somewhat stronger bond. However, different bonding types also lead to different material properties, which may account for or contribute to the optical performance. One important property is the Tg of the resulting system, and, for the comparisons made, the Tg of the ionic complexes is much higher than that of the hydrogen-bonded complexes. Similarly, the Tg of the halogen-bonded complex was higher than that of the analogous hydrogen-bonded complex. In these comparisons, the polymer chain length is constant (since the same polymer is used), but the question of how the molecular weight, including in a range where the Tg is essentially constant, might affect optical performances is also of interest in determining the efficiency of the different supramolecular bond types.
The best performing photoinduced anisotropy materials generally have liquid-crystalline character, which can promote cooperative molecular motion and thereby possibly drive more efficient photo-orientation and photo-induced surface patterning. Although it is hard to imagine a perfect comparison between an LC and a non-LC material, since it involves modifying the chemical structure, maybe the closest current example, enabled by supramolecular design, is a gradual variation of the azobenzene content over a critical range where the structure of the material adopts LC phase (analogously to lyotropic liquid crystals). Studies done with hydrogen-bonded complexes unanimously point in the direction that the introduction of an LC phase by adding more azobenzenes not only increases the overall photo-orientation and its temporal stability, but also the contribution per chromophore in the system to the total orientation. However, the picture drawn for the relationship of liquid crystallinity and SRG formation is not yet settled, since isotropic hydrogen-bonded materials sometimes outperform the liquid-crystalline ones, and both highly efficient liquid-crystalline and non-liquid-crystalline materials have been reported.
Whether the strength of the supramolecular interaction needs to be augmented or decreased for better performance is an intriguing question. Overall, the ionic bonds studied thus far are at least an order of magnitude stronger than the studied hydrogen and halogen bonds. Taking into account the general good performance of the reviewed ionic complexes, as well as the direct correlation of increasing halogen-bond strength and increased efficiency in photoresponse, the field would benefit from studies designing stronger hydrogen and halogen bonds than have been studied to date. However, using less strong interactions might be beneficial, when preparing complexes of very low azobenzene contents, since the dynamic character of hydrogen and halogen bonding may allow hopping of the azobenzenes between different polymer repeat units, which is hypothesized to increase surface-relief formation efficiency. To study this hypothesis, libraries of gradually varying azobenzene contents of ionically-bonded complexes should be prepared and compared to existing hydrogen- and halogen-bonded materials. This is of interest, since using very low azobenzene contents would allow using thicker films for photo-orientation or (semi)-transparent films.
Completing the understanding of parameters that make or break the performance of a material system would definitely be easier, if the efforts towards that goal would be less scattered. When revisiting the reviewed literature, the reader is suggested to keep in mind that the photoresponse of a material is influenced by various experimental parameters such as the film preparation, ambient humidity, laser wavelength, and intensity. Thus a reliable and straightforward comparison between experimental results for different materials, even for the same material made by different groups or at different times of the year, is rarely possible. Setting benchmarks of the performance needed for applications benefiting from photo-orientation are also required to facilitate comparisons between the different materials.
During the last 10 years, the materials science community has been able to demonstrate that supramolecular polymer–azobenzene complexes can reach and even surpass the performance of conventional covalent side-chain azopolymers. A question of critical importance is what is needed to bring this research to the next level, whether it means taking one step towards applications or a design that would enable a breakthrough in materials performance? Many of the reviewed relatively simple material designs can be regarded as model systems for more complicated host molecule architectures. One step at a time, we believe that the complexity of these materials will be increased. Many cross-disciplinary fields would benefit from a similar kind of simple and systematic materials design approach for establishing design guidelines for desired applications. Given the facility and versatility of available supramolecular approaches, there is room for gathering additional fundamental understanding and then optimizing the materials for a wealth of applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
JV acknowledges funding from the Academy of Finland and a Banting Postdoctoral Fellowship (Canada). AP is thankful for the Academy of Finland (decision numbers 277091 and 312628) and the Emil Aaltonen Foundation for financial support. GB acknowledges support from the Natural Sciences and Engineering Research Council (NSERC) of Canada and the Fonds de Recherche du Québec - Nature et Technologies (FRQNT).Notes and references
- J.-M. Lehn, Angew. Chem., 1988, 100, 91–116 CrossRef CAS.
- J.-M. Lehn, Acc. Chem. Res., 1978, 11, 49–57 CrossRef CAS.
- J.-M. Lehn, Pure Appl. Chem., 1978, 50, 871–892 CrossRef CAS.
- J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, Wiley-VCH (Verlag), Weinheim, Germany, 1995 Search PubMed.
- J. Steed and J. Atwood, Supramolecular Chemistry, John Wiley & Sons Ltd., 2000 Search PubMed.
- C. F. J. Faul and M. Antonietti, Adv. Mater., 2003, 15, 673–683 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- M. Nič, J. Jirát, B. Košata, A. Jenkins and A. McNaught, IUPAC. Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”), Blackwell Scientific Publications, Oxford, 1997 Search PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- P. A. Kollman and L. C. Allen, Chem. Rev., 1972, 72, 283–303 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32 CrossRef CAS.
- H. M. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- D. Bleger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef CAS PubMed.
- J. de Wit, G. A. van Ekenstein, E. Polushkin, K. Kvashnina, W. Bras, O. Ikkala and G. ten Brinke, Macromolecules, 2008, 41, 4200–4204 CrossRef CAS.
- X. Sallenave and C. G. Bazuin, Macromolecules, 2007, 40, 5326–5336 CrossRef CAS.
- T. Kato, N. Hirota, A. Fujishima and J. M. J. Frechet, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 57–62 CrossRef CAS.
- S. Ujiie and K. Iimura, Macromolecules, 1992, 25, 3174–3178 CrossRef CAS.
- M. Poutanen, O. Ikkala and A. Priimagi, Macromolecules, 2016, 49, 4095–4101 CrossRef CAS.
- A. Priimagi, A. Shevchenko, M. Kaivola, F. J. Rodriguez, M. Kauranen and P. Rochon, Opt. Lett., 2010, 35, 1813–1815 CrossRef CAS PubMed.
- M. Utecht, T. Klamroth and P. Saalfrank, Phys. Chem. Chem. Phys., 2011, 13, 21608–21614 RSC.
- F. Weigert, Z. Phys. Chem., 1929, 3, 377–388 CAS.
- A. M. Makushenko, B. S. Neporent and O. V. Stolbova, Opt. Spectrosc., 1971, 31, 295–299 Search PubMed.
- A. M. Makushenko, B. S. Neporent and O. V. Stolbova, Opt. Spectrosc., 1971, 31, 741–749 CAS.
- T. Todorov, L. Nikolova and N. Tomova, Appl. Opt., 1984, 23, 4588–4591 CrossRef CAS PubMed.
- K. Anderle, R. Birenheide, M. Eich and J. H. Wendorff, Makromol. Chem., Rapid Commun., 1989, 10, 477–483 CrossRef CAS.
- M. Eich, J. H. Wendorff, B. Reck and H. Ringsdorf, Makromol. Chem., Rapid Commun., 1987, 8, 59–63 CrossRef CAS.
- G. S. Kumar and D. C. Neckers, Chem. Rev., 1989, 89, 1915–1925 CrossRef CAS.
- S. Xie, A. Natansohn and P. Rochon, Chem. Mater., 1993, 5, 403–411 CrossRef CAS.
- D. Brown, A. Natansohn and P. Rochon, Macromolecules, 1995, 28, 6116–6123 CrossRef CAS.
- A. Natansohn, P. Rochon, J. Gosselin and S. Xie, Macromolecules, 1992, 25, 2268–2273 CrossRef CAS.
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139–4176 CrossRef CAS PubMed.
- D. Y. Kim, S. K. Tripathy, L. Li and J. Kumar, Appl. Phys. Lett., 1995, 66, 1166–1168 CrossRef CAS.
- P. Rochon, E. Batalla and A. Natansohn, Appl. Phys. Lett., 1995, 66, 136–138 CrossRef CAS.
- A. Priimagi, M. Kaivola, M. Virkki, F. J. Rodríguez and M. Kauranen, J. Nonlinear Opt. Phys. Mater., 2010, 19, 57–73 CrossRef CAS.
- M. Saphiannikova, W. Toshchevikov and J. Ilnytskyi, Nonlinear Opt., Quantum Opt., 2010, 41, 27–57 CAS.
- M. L. Juan, J. Plain, R. Bachelot, P. Royer, S. K. Gray and G. P. Wiederrecht, ACS Nano, 2009, 3, 1573–1579 CrossRef CAS PubMed.
- C. F. P. Lefin and J.-M. Nunzi, Pure Appl. Opt., 1998, 7, 71–82 CrossRef.
- S. Ciobotarescu, N. Hurduc and V. Teboul, Phys. Chem. Chem. Phys., 2016, 18, 14654–14661 RSC.
- A. Ambrosio, L. Marrucci, F. Borbone, A. Roviello and P. Maddalena, Nat. Commun., 2012, 3, 989 CrossRef PubMed.
- F. Fabbri, Y. Lassailly, S. Monaco, K. Lahlil, J. P. Boilot and J. Peretti, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 115440 CrossRef.
- J. Isayama, S. Nagano and T. Seki, Macromolecules, 2010, 43, 4105–4112 CrossRef CAS.
- T. Seki, Macromol. Rapid Commun., 2014, 35, 271–290 CrossRef CAS PubMed.
- N. Zettsu, T. Ogasawara, R. Arakawa, S. Nagano, T. Ubukata and T. Seki, Macromolecules, 2007, 40, 4607–4613 CrossRef CAS.
- N. C. R. Holme, L. Nikolova, P. S. Ramanujam and S. Hvilsted, Appl. Phys. Lett., 1997, 70, 1518–1520 CrossRef CAS.
- A. Sobolewska and A. Miniewicz, J. Phys. Chem. B, 2007, 111, 1536–1544 CrossRef CAS PubMed.
- N. K. Viswanathan, D. Y. Kim, S. Bian, J. Williams, W. Liu, L. Li, L. Samuelson, J. Kumar and S. K. Tripathy, J. Mater. Chem., 1999, 9, 1941–1955 RSC.
- V. Shibaev, A. Bobrovsky and N. Boiko, Prog. Polym. Sci., 2003, 28, 729–836 CrossRef CAS.
- O. N. Oliveira, Jr., D. S. Dos Santos, Jr., D. T. Balogh, V. Zucolotto and C. R. Mendonca, Adv. Colloid Interface Sci., 2005, 116, 179–192 CrossRef PubMed.
- R. C. Advincula, in Handbook of Polyelectrolytes and Their Applications, ed. S. K. Tripathy, J. Kumar and H. S. Nalwa, American Scientific Publishers, Stevenson Ranch, CA, 2002, pp. 65–97 Search PubMed.
- J. Stumpe, O. Kulikovska, L. M. Goldenberg and Y. Zakrevskyy, in Smart Light-Responsive Materials, ed. Y. Zhao and T. Ikeda, John Wiley & Sons Ltd., Hoboken, NJ, USA, 2009, pp. 47–94 Search PubMed.
- C. F. Faul, Acc. Chem. Res., 2014, 47, 3428–3438 CrossRef CAS PubMed.
- T. Ube and T. Ikeda, Angew. Chem., Int. Ed., 2014, 53, 10290–10299 CrossRef CAS PubMed.
- H. Yu, J. Mater. Chem. C, 2014, 2, 3047–3054 RSC.
- H. Yu, Prog. Polym. Sci., 2014, 39, 781–815 CrossRef CAS.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- M. Saccone, G. Cavallo, P. Metrangolo, G. Resnati and A. Priimagi, Top. Curr. Chem., 2015, 359, 147–166 CrossRef CAS PubMed.
- Y. Zhao and J. He, Soft Matter, 2009, 5, 2686–2693 RSC.
- M. Hendrikx, A. Schenning, M. Debije and D. Broer, Crystals, 2017, 7, 231 CrossRef.
- M. Baroncini and G. Bergamini, Chem. Rec., 2017, 17, 700–712 CrossRef CAS PubMed.
- O. S. Bushuyev, M. Aizawa, A. Shishido and C. J. Barrett, Macromol. Rapid Commun., 2018, 39, 1700253 CrossRef PubMed.
- S. Santer, J. Phys. D: Appl. Phys., 2018, 51, 013002 CrossRef.
- K. Ichimura, in Polymers as Electrooptical and Photooptical Active Media, ed. V. P. Shibaev, Springer, Berlin, Germany, 1996, pp. 138–172 Search PubMed.
- S. Lee, H. S. Kang and J. K. Park, Adv. Mater., 2012, 24, 2069–2103 CrossRef CAS PubMed.
- T. Seki, K. Fukuhara, M. Sano, M. Hara and S. Nagano, Mol. Cryst. Liq. Cryst., 2015, 614, 118–123 CrossRef CAS.
- A. Natansohn and P. Rochon, Adv. Mater., 1999, 11, 1387–1391 CrossRef CAS.
- A. Priimagi and A. Shevchenko, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 163–182 CrossRef CAS.
- R. Ahmed, A. Priimagi, C. F. J. Faul and I. Manners, Adv. Mater., 2012, 24, 926–931 CrossRef CAS PubMed.
- C. Frenz, A. Fuchs, H.-W. Schmidt, U. Theissen and D. Haarer, Macromol. Chem. Phys., 2004, 205, 1246–1258 CrossRef CAS.
- S.-C. Tsao and C.-T. Lo, RSC Adv., 2014, 4, 23585–23594 RSC.
- K. Moriya, T. Seki, M. Nakagawa, G. Mao and C. K. Ober, Macromol. Rapid Commun., 2000, 21, 1309–1312 CrossRef CAS.
- H. Yu, K. Okano, A. Shishido, T. Ikeda, K. Kamata, M. Komura and T. Iyoda, Adv. Mater., 2005, 17, 2184–2188 CrossRef CAS.
- A. M. Sweetman, S. P. Jarvis, H. Sang, I. Lekkas, P. Rahe, Y. Wang, J. Wang, N. R. Champness, L. Kantorovich and P. Moriarty, Nat. Commun., 2014, 5, 3931 CAS.
- W. O. George and R. Lewis, Hydrogen Bonding. Handbook of Vibrational Spectroscopy, John Wiley & Sons, Ltd, 2006 Search PubMed.
- J. Vapaavuori, C. G. Bazuin and C. Pellerin, Macromol. Rapid Commun., 2018, 39, 1700430 CrossRef PubMed.
- X. Wang, C. G. Bazuin and C. Pellerin, Vib. Spectrosc., 2014, 71, 18–23 CrossRef CAS.
- F. Wang, N. Ma, Q. Chen, W. Wang and L. Wang, Langmuir, 2007, 23, 9540–9542 CrossRef CAS PubMed.
- J. Xu, X. Liu, J. K.-P. Ng, T. Lin and C. He, J. Mater. Chem., 2006, 16, 3540 RSC.
- B. Fortier-McGill, V. Toader and L. Reven, Macromolecules, 2012, 45, 6015–6026 CrossRef CAS.
- Q. Zhang and C. G. Bazuin, Macromolecules, 2009, 42, 4775–4786 CrossRef CAS.
- Q. Zhang, X. Wang, C. J. Barrett and C. G. Bazuin, Chem. Mater., 2009, 21, 3216–3227 CrossRef CAS.
- X. Wang, J. Vapaavuori, X. Wang, R. G. Sabat, C. Pellerin and C. G. Bazuin, Macromolecules, 2016, 49, 4923–4934 CrossRef CAS.
- J. Vapaavuori, I. T. S. Heikkinen, V. Dichiarante, G. Resnati, P. Metrangolo, R. G. Sabat, C. G. Bazuin, A. Priimagi and C. Pellerin, Macromolecules, 2015, 48, 7535–7542 CrossRef CAS.
- S. Schoelch, J. Vapaavuori, F. G. Rollet and C. J. Barrett, Macromol. Rapid Commun., 2017, 38, 1600582 CrossRef PubMed.
- L. Shi, X. Ran, Y. Li, Q. Li, W. Qiu and L. Guo, RSC Adv., 2015, 5, 38283–38289 RSC.
- H. Ren, D. Chen, Y. Shi, H. Yu and Z. Fu, Polym. Chem., 2015, 6, 270–277 RSC.
- H. Ren, D. Chen, Y. Shi, H. Yu and Z. Fu, Polymer, 2016, 97, 533–542 CrossRef CAS.
- A. Priimagi, S. Cattaneo, R. H. A. Ras, S. Valkama, O. Ikkala and M. Kauranen, Chem. Mater., 2005, 17, 5798–5802 CrossRef CAS.
- A. Priimagi, M. Kaivola, F. J. Rodriguez and M. Kauranen, Appl. Phys. Lett., 2007, 90, 121103 CrossRef.
- A. Priimagi, J. Vapaavuori, F. J. Rodriguez, C. F. J. Faul, M. T. Heino, O. Ikkala, M. Kauranen and M. Kaivola, Chem. Mater., 2008, 20, 6358–6363 CrossRef CAS.
- C. G. Bazuin and A. Tork, Macromolecules, 1995, 28, 8877–8880 CrossRef CAS.
- Q. Zhang, C. G. Bazuin and C. J. Barrett, Chem. Mater., 2008, 20, 29–31 CrossRef CAS.
- J. Vapaavuori, V. Valtavirta, T. Alasaarela, J.-I. Mamiya, A. Priimagi, A. Shishido and M. Kaivola, J. Mater. Chem., 2011, 21, 15437–15441 RSC.
- M. J. Banach, M. D. Alexander, S. Caracci and R. A. Vaia, Chem. Mater., 1999, 11, 2554–2561 CrossRef CAS.
- S. Wu and C. Bubeck, Macromolecules, 2013, 46, 3512–3518 CrossRef CAS.
- N. Zettsu, T. Ogasawara, N. Mizoshita, S. Nagano and T. Seki, Adv. Mater., 2008, 20, 516–521 CrossRef CAS.
- A. V. Medvedev, E. B. Barmatov, A. S. Medvedev, V. P. Shibaev, S. A. Ivanov, M. Kozlovsky and J. Stumpe, Macromolecules, 2005, 38, 2223–2229 CrossRef CAS.
- L. Cui and Y. Zhao, Chem. Mater., 2004, 16, 2076–2082 CrossRef CAS.
- Y. Zakrevskyy, J. Stumpe and C. F. J. Faul, Adv. Mater., 2006, 18, 2133–2136 CrossRef CAS.
- Y. Zakrevskyy, J. Stumpe, B. Smarsly and C. F. Faul, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 75, 031703 CrossRef CAS PubMed.
- S. Xiao, X. Lu and Q. Lu, Macromolecules, 2007, 40, 7944–7950 CrossRef.
- X. Pan, S. Xiao, C. Wang, P. Cai, X. Lu and Q. Lu, Opt. Commun., 2009, 282, 763–768 CrossRef CAS.
- K. Kreger, P. Wolfer, H. Audorff, L. Kador, N. Stingelin-Stutzmann, P. Smith and H. W. Schmidt, J. Am. Chem. Soc., 2010, 132, 509–516 CrossRef CAS PubMed.
- M. Marcos, R. Alcalá, J. Barberá, P. Romero, C. Sánchez and J. L. Serrano, Chem. Mater., 2008, 20, 5209–5217 CrossRef CAS.
- S. Hernández-Ainsa, R. Alcalá, J. Barberá, M. Marcos, C. Sánchez and J. L. Serrano, Eur. Polym. J., 2011, 47, 311–318 CrossRef.
- S. Hernández-Ainsa, R. Alcalá, J. n. Barberá, M. Marcos, C. Sánchez and J. L. Serrano, Macromolecules, 2010, 43, 2660–2663 CrossRef.
- J. Gao, Y. He, H. Xu, B. Song, X. Zhang, Z. Wang and X. Wang, Chem. Mater., 2007, 19, 14–17 CrossRef CAS.
- J. Gao, Y. He, F. Liu, X. Zhang, Z. Wang and X. Wang, Chem. Mater., 2007, 19, 3877–3881 CrossRef CAS.
- C. Fiorini, F. Charra, J.-M. Nunzi and P. Raimond, J. Opt. Soc. Am. B, 1997, 14, 1984 CrossRef.
- M. Virkki, M. Kauranen and A. Priimagi, Appl. Phys. Lett., 2011, 99, 183309 CrossRef.
- J. E. Koskela, J. Vapaavuori, J. Hautala, A. Priimagi, C. F. J. Faul, M. Kaivola and R. H. A. Ras, J. Phys. Chem. C, 2012, 116, 2363–2370 CAS.
- J. E. Koskela, J. Vapaavuori, R. H. A. Ras and A. Priimagi, ACS Macro Lett., 2014, 3, 1196–1200 CrossRef CAS.
- J. Vapaavuori, A. Priimagi and M. Kaivola, J. Mater. Chem., 2010, 20, 5260–5264 RSC.
- J. Vapaavuori, A. Goulet-Hanssens, I. T. S. Heikkinen, C. J. Barrett and A. Priimagi, Chem. Mater., 2014, 26, 5089–5096 CrossRef CAS.
- S. Wu, S. Duan, Z. Lei, W. Su, Z. Zhang, K. Wang and Q. Zhang, J. Mater. Chem., 2010, 20, 5202–5209 RSC.
- X. Wang, J. Vapaavuori, C. G. Bazuin and C. Pellerin, Macromolecules, 2018 DOI:10.1021/acs.macromol.7b02534.
- M. Hackel, L. Kador, D. Kropp and H. W. Schmidt, Adv. Mater., 2007, 19, 227–231 CrossRef.
- A. Saishoji, D. Sato, A. Shishido and T. Ikeda, Langmuir, 2007, 23, 320–326 CrossRef CAS PubMed.
- J. del Barrio, E. Blasco, L. Oriol, R. Alcalá and C. Sánchez-Somolinos, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1716–1725 CrossRef CAS.
- J. del Barrio, E. Blasco, C. Toprakcioglu, A. Koutsioubas, O. A. Scherman, L. Oriol and C. Sánchez-Somolinos, Macromolecules, 2014, 47, 897–906 CrossRef CAS.
- A. Concellón, E. Blasco, M. Piñol, L. Oriol, I. Díez, C. Berges, C. Sánchez-Somolinos and R. Alcalá, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3173–3184 CrossRef.
- A. Priimagi, M. Saccone, G. Cavallo, A. Shishido, T. Pilati, P. Metrangolo and G. Resnati, Adv. Mater., 2012, 24, OP345–OP352 CAS.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- F. Fernandez-Palacio, M. Poutanen, M. Saccone, A. Siiskonen, G. Terraneo, G. Resnati, O. Ikkala, P. Metrangolo and A. Priimagi, Chem. Mater., 2016, 28, 8314–8321 CrossRef CAS PubMed.
- J. Vapaavuori, A. Siiskonen, V. Dichiarante, A. Forni, M. Saccone, T. Pilati, C. Pellerin, A. Shishido, P. Metrangolo and A. Priimagi, RSC Adv., 2017, 7, 40237–40242 RSC.
- Y. Liang, D. Mauran, R. E. Prud'homme and C. Pellerin, Appl. Spectrosc., 2008, 62, 941–947 CrossRef CAS PubMed.
- M. Virkki, O. Tuominen, A. Forni, M. Saccone, P. Metrangolo, G. Resnati, M. Kauranen and A. Priimagi, J. Mater. Chem. C, 2015, 3, 3003–3006 RSC.
- O. Kulikovska, L. M. Goldenberg and J. Stumpe, Chem. Mater., 2007, 19, 3343–3348 CrossRef CAS.
- F. Lagugné Labarthet, T. Buffeteau and C. Sourisseau, J. Phys. Chem. B, 1998, 102, 2654–2662 CrossRef.
- O. Kulikovska, L. M. Goldenberg, L. Kulikovsky and J. Stumpe, Chem. Mater., 2008, 20, 3528–3534 CrossRef CAS.
- T. König, L. M. Goldenberg, O. Kulikovska, L. Kulikovsky, J. Stumpe and S. Santer, Soft Matter, 2011, 7, 4174–4178 RSC.
- N. S. Yadavalli, T. Konig and S. Santer, J. Soc. Inf. Disp., 2015, 23, 154–162 CrossRef CAS.
- J. E. Koskela, V. Liljestrom, J. Lim, E. E. Simanek, R. H. Ras, A. Priimagi and M. A. Kostiainen, J. Am. Chem. Soc., 2014, 136, 6850–6853 CrossRef CAS PubMed.
- J. Vapaavuori, A. Priimagi, A. J. Soininen, N. Canilho, E. Kasëmi, J. Ruokolainen, M. Kaivola and O. Ikkala, Opt. Mater. Express, 2013, 3, 711–722 CrossRef CAS.
- F. Zeng and S. C. Zimmerman, Chem. Rev., 1997, 97, 1681–1712 CrossRef CAS PubMed.
- A. Priimagi, K. Lindfors, M. Kaivola and P. Rochon, ACS Appl. Mater. Interfaces, 2009, 1, 1183–1189 CAS.
- J. Vapaavuori, R. H. A. Ras, M. Kaivola, C. G. Bazuin and A. Priimagi, J. Mater. Chem. C, 2015, 3, 11011–11016 RSC.
- A. Sobolewska and S. Bartkiewicz, Appl. Phys. Lett., 2012, 101, 193301 CrossRef.
- J. Noga, A. Sobolewska, S. Bartkiewicz, M. Virkki and A. Priimagi, Macromol. Mater. Eng., 2017, 302, 1600329 CrossRef.
- A. Sobolewska and S. Bartkiewicz, J. Mater. Chem. C, 2015, 3, 5616–5620 RSC.
- E. Schab-Balcerzak, H. Flakus, A. Jarczyk-Jedryka, J. Konieczkowska, M. Siwy, K. Bijak, A. Sobolewska and J. Stumpe, Opt. Mater., 2015, 47, 501–511 CrossRef CAS.
- E. Schab-Balcerzak, J. Konieczkowska, M. Siwy, A. Sobolewska, M. Wojtowicz and M. Wiacek, Opt. Mater., 2014, 36, 892–902 CrossRef CAS.
- A. Priimagi, G. Cavallo, A. Forni, M. Gorynsztejn-Leben, M. Kaivola, P. Metrangolo, R. Milani, A. Shishido, T. Pilati, G. Resnati and G. Terraneo, Adv. Funct. Mater., 2012, 22, 2572–2579 CrossRef CAS.
- M. Saccone, V. Dichiarante, A. Forni, A. Goulet-Hanssens, G. Cavallo, J. Vapaavuori, G. Terraneo, C. J. Barrett, G. Resnati, P. Metrangolo and A. Priimagi, J. Mater. Chem. C, 2015, 3, 759–768 RSC.
- J. E. Stumpel, M. Saccone, V. Dichiarante, O. Lehtonen, M. Virkki, P. Metrangolo and A. Priimagi, Molecules, 2017, 22, 1844 CrossRef PubMed.
- O. Ikkala and G. ten Brinke, Science, 2002, 295, 2407–2409 CrossRef CAS PubMed.
- Y. Zhao, K. Thorkelsson, A. J. Mastroianni, T. Schilling, J. M. Luther, B. J. Rancatore, K. Matsunaga, H. Jinnai, Y. Wu, D. Poulsen, J. M. Frechet, A. P. Alivisatos and T. Xu, Nat. Mater., 2009, 8, 979–985 CrossRef CAS PubMed.
- A. Sidorenko, I. Tokarev, S. Minko and M. Stamm, J. Am. Chem. Soc., 2003, 125, 12211–12216 CrossRef CAS PubMed.
- B. Nandan, M. K. Vyas, M. Böhme and M. Stamm, Macromolecules, 2010, 43, 2463–2473 CrossRef CAS.
- B. Nandan, E. B. Gowd, N. C. Bigall, A. Eychmüller, P. Formanek, P. Simon and M. Stamm, Adv. Funct. Mater., 2009, 19, 2805–2811 CrossRef CAS.
- P. Hiekkataipale, T. I. Löbling, M. Poutanen, A. Priimagi, V. Abetz, O. Ikkala and A. H. Gröschel, Polymer, 2016, 107, 456–465 CrossRef CAS.
- Y. Morikawa, S. Nagano, K. Watanabe, K. Kamata, T. Iyoda and T. Seki, Adv. Mater., 2006, 18, 883–886 CrossRef CAS.
- T. Seki, J. Mater. Chem. C, 2016, 4, 7895–7910 RSC.
- A. Kopyshev, C. J. Galvin, J. Genzer, N. Lomadze and S. Santer, Langmuir, 2013, 29, 13967–13974 CrossRef CAS PubMed.
- J. Vapaavuori, J. Grosrenaud, C. Pellerin and C. G. Bazuin, ACS Macro Lett., 2015, 4, 1158–1162 CrossRef CAS.
- S. Roland, C. G. Gamys, J. Grosrenaud, S. Boissé, C. Pellerin, R. E. Prud’homme and C. G. Bazuin, Macromolecules, 2015, 48, 4823–4834 CrossRef CAS.
- S. Roland, R. E. Prud’homme and C. G. Bazuin, ACS Macro Lett., 2012, 1, 973–976 CrossRef CAS.
- H. Yu, H. Liu and T. Kobayashi, ACS Appl. Mater. Interfaces, 2011, 3, 1333–1340 CAS.
- H. Liu, T. Kobayashi and H. Yu, Macromol. Rapid Commun., 2011, 32, 378–383 CrossRef CAS PubMed.
- H. Zhang, R. Hao, J. K. Jackson, M. Chiao and H. Yu, Chem. Commun., 2014, 50, 14843–14846 RSC.
- D. Chen, H. Liu, T. Kobayashi and H. Yu, J. Mater. Chem., 2010, 20, 3610–3614 RSC.
- S. Wu, J. Huang, S. Beckemper, A. Gillner, K. Wang and C. Bubeck, J. Mater. Chem., 2012, 22, 4989 RSC.
- T. Alasaarela, D. Zheng, L. Huang, A. Priimagi, B. Bai, A. Tervonen, S. Honkanen, M. Kuittinen and J. Turunen, Opt. Lett., 2011, 36, 2411–2413 CrossRef PubMed.
- T. Ubukata, T. Isoshima and M. Hara, Adv. Mater., 2005, 1630–1633 CrossRef CAS.
- L. M. Goldenberg, V. Lisinetskii and S. Schrader, Adv. Opt. Mater., 2013, 1, 527–533 CrossRef.
- S. L. Lim, N.-J. Li, J.-M. Lu, Q.-D. Ling, C. X. Zhu, E.-T. Kang and K. G. Neoh, ACS Appl. Mater. Interfaces, 2009, 1, 60–71 CAS.
- T. Mosciatti, S. Bonacchi, M. Gobbi, L. Ferlauto, L. Fabiola, L. Giorgini, E. Orgiu and P. Samori, ACS Appl. Mater. Interfaces, 2016, 8, 6563–6569 CAS.
- D. Attianese, M. Petrosino, P. Vacca, S. Concilio, P. Iannelli, A. Rubino and B. Salvatore, IEEE Electron Device Lett., 2008, 29, 44–46 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |