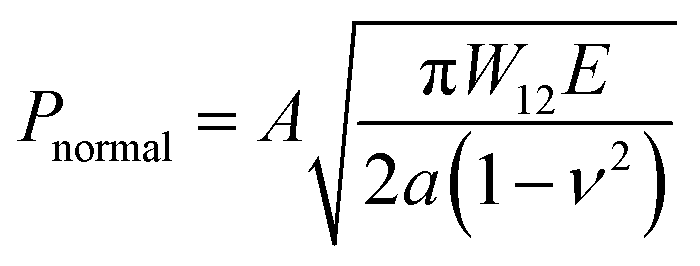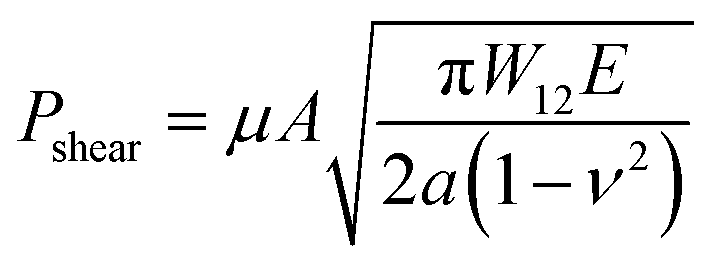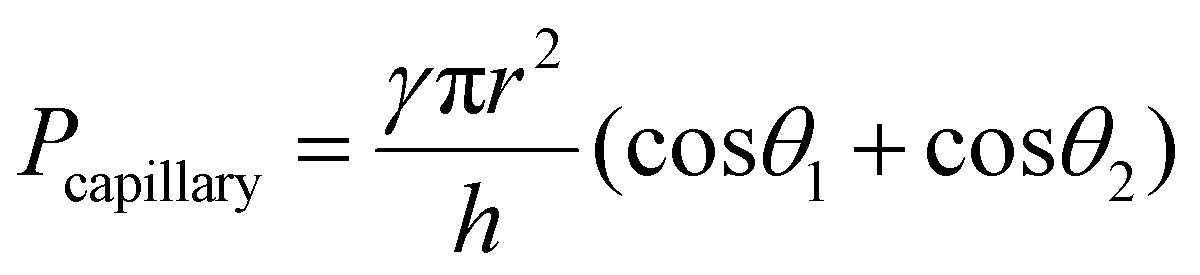Bioinspired reversible hydrogel adhesives for wet and underwater surfaces†
Received
2nd October 2018
, Accepted 6th November 2018
First published on 6th November 2018
Abstract
Stable and reversible adhesion to wet surfaces is challenging owing to water molecules at the contact interface. In this study, we develop a hydrogel-based wet adhesive, which can exhibit strong and reversible adhesion to wet and underwater surfaces as well as to dry surfaces. The remarkable wet adhesion of the hydrogel adhesive is realized based on a synergetic integration of bioinspired microarchitectures and water-friendly and water-absorbing properties of the polymeric hydrogel. Under dry conditions, the microstructured hydrogel adhesive exhibits strong van der Waals interaction-based adhesion, while under underwater conditions, it can maximize capillary adhesion. Consequently, the hydrogel adhesive exhibits remarkable adhesion strengths for dry, moist, and submerged substrates. Maximum normal and shear adhesion strengths of 423 and 384, 492 and 340, and 253 and 21 kPa are achieved with the hydrogel adhesive for dry, moist, and submerged substrates, respectively. Our results demonstrate that strong wet and underwater adhesion can be achieved only with the hydrogel-based adhesive with simple microscale architecture.
Introduction
Robust adhesion to wet surfaces is of significance in diverse applications such as medical operations, marine structures, energy devices operating under wet environments (e.g., fuel cells), transfer printing, and even daily goods.1–8 Stable adhesion between surfaces can be obtained by keeping the distance between the surfaces very close to the atomic level. In typical dry environments, such tight contact between surfaces can be easily achieved by utilizing materials with low effective elastic moduli so that they can follow the roughness of the target surface.9–17 However, in wet environments, water molecules not only penetrate gaps between the surfaces easily, but also reduce the adhesion strengths of the adhesive materials by hydrating and decomposing them. Consequently, it is very challenging to ensure good adhesion under wet conditions1,3,8,18–20 Although cyanoacrylate (Super Glue™) is a strong tissue adhesive, it is cytotoxic, and incompatible with wet or underwater surfaces because the uncured adhesive rapidly solidifies into a glassy phase when it comes into contact with water.21,22 In order to overcome such difficulties, synthetic wet adhesives emulating natural adhesive materials of marine organisms have been extensively investigated.23 Representative examples include wet adhesives mimicking the proteins present in biological system such as mussel adhesives,23–26 barnacle cements,27 and sandcastle-worm glues.28 These marine organism-inspired synthetic wet adhesives typically have the use of 3,4-dihydroxyphenylalanine (DOPA) in common, and many different types of DOPA-based wet adhesives have demonstrated effective wet adhesion performance.1,5,7,29 However, they also pose several limitations such as adhesion degradation by wet-air oxidation, long contact-forming time, and high production cost. In addition, they are not suitable for applications that require reversible and reusable wet adhesion, as they are typically permanent adhesives that lack reversible adhesion capability1,30
Bioinspired dry adhesives based on micro- or nanostructure arrays that emulate biological fibrillar adhesives have emerged as reversible and reusable adhesives for various dry surfaces. Their reversible dry adhesion could be realized by maximizing van der Waals interactions with the target surface with the aid of the micro- or nanostructure array.31–41 Most of the existing bioinspired dry adhesives are composed of elastomers with hydrophobic surface properties (e.g., polydimethylsiloxane (PDMS)).42–49 This indicates that they have potential as wet or underwater adhesives because hydrophobic elastomers promote removal of the water at the contact interface and therefore initiate interfacial adhesion in wet environments.50,51 In reality, however, water entrapment often occurs at the contact interface even when using a hydrophobic elastomer because it is difficult to perfectly avoid or remove any entrapped water across the entire surface of elastomeric samples of macroscopic size.52 In this case, the entrapped water acts as a flaw for initiating debonding at the interface,52 resulting in limited wet adhesion performance.1,3,8,18–20 In this regard, hydrogels have immense potential as reversible wet adhesives when they are integrated with biomimetic microstructures, based on the following factors. First, the hydrophilic nature of the hydrogels is beneficial to induce strong capillary adhesion under wet conditions. Second, they can absorb a large quantity of water at the contact interface, which would allow closer contact with the target substrate.3,19,53–55 Furthermore, they are biocompatible and therefore can be used for diverse biomedical applications such as tissue adhesives, tissue repair, and wound dressings. Indeed, in recent years, hydrogels have been actively explored as materials for efficient wet adhesives.21,22,52,56 Engineered tough hydrogel adhesives have been demonstrated to have remarkable adhesion performance with an adhesion energy over 1000 J m−2 to various substrates including skin, tissue, glass, and metals.21,22,56 However, they also have several shortcomings such as requirements for specific functional groups, long contact-forming time, and irreversible adhesion.21,22,52,56 To address the shortcomings, hydrogel adhesives with unique micro- and nano-structures have been used, and their fast and reversible underwater adhesion performance has been demonstrated based on ionic and hydrogen bonds, mechanical interlocking, and pH-sensitive swelling.3,52,57 Nonetheless, the research toward reversible, fast, and strong adhesives for wet and underwater surfaces is still in its infancy and requires further development.52
In this study, we develop a bioinspired wet adhesive utilizing a hydrogel, which can exhibit immediate, strong and reversible adhesion under wet and underwater conditions based on the water-absorbing properties and capillary adhesion of the microstructured hydrogel. The hydrogel adhesive consists of a bioinspired micropillar array made of the biocompatible polyethylene glycol (PEG) hydrogel, which can absorb a large quantity of water and is hydrophilic. In this PEG-based bioinspired adhesive, the biomimetic microarchitecture enables conformal physical contact to target surfaces, while the PEG polymer absorbs water at the contact interface and enables capillary adhesion. Consequently, robust and reversible wet adhesion can be achieved under wet and underwater conditions only with the hydrogel adhesive, without the need for complex surface modification with chemical moieties. The wet adhesion behavior of the bioinspired hydrogel adhesive well agrees with theoretical models.
Experimental methods
Fabrication of the hydrogel adhesive
A master mold with a microhole array was prepared by photolithography using a bilayer of a lift-off resist (LOR, Microchem Corporation) and SU-8 photoresist. First, the LOR was spin-coated onto a Si wafer followed by baking at 200 °C for 10 min. SU-8 was then spin-coated onto the LOR layer and baked at 100 °C for 5 min. Subsequently, the SU-8/LOR bilayer on the wafer was exposed to ultraviolet (UV) light (λ = 365 nm, dose = 250 mJ cm−2), followed by post-exposure baking (100 °C for 3 min). Microhole array patterns were then generated in the SU-8 layer by development using a SU-8 developer. Subsequently, selective removal of the LOR layer was carried out to form an undercut in the LOR layer using a LOR developer (AZ 400K, AZ Electronics Materials Corporation). In order to generate a PEG dimethacrylate (PEGDMA) adhesive, a drop of an 80 wt% PEGDMA solution (molecular weight: 750, Sigma Aldrich) mixed with 20 wt% of deionized (DI) water and 0.2 wt% of an Irgacure 2959 photoinitiator (Sigma Aldrich) was uniformly dispensed onto the patterned master.58–61 Subsequent photopolymerization of the PEGDMA solution by UV irradiation (λ = 365 nm, dose = 300 mJ cm−2) and removal of the cured PEGDMA from the master yielded the PEGDMA adhesive with a bioinspired microstructure array.62,63
Surface characterization
Scanning electron microscopy (SEM) images of the fabricated PEGDMA sample were acquired using a HITACHI S-4800 microscope (Hitachi, Japan). To image the swollen PEGDMA microstructures, the PEGDMA sample was first submerged in DI water for 12 h. Next, the swollen PEGDMA sample was lyophilized. In order to avoid charging effects, the PEGDMA sample was coated with a 5 nm-thick Pt layer using a sputter coater (K575X, Quorum Emitech, UK). The water contact angles (CAs) on the planar PEGDMA and glass substrates were measured using a drop shape analyzer (SDLAB 200TEZD, FEMTOFAB, Korea) at room temperature (25 °C). The measurements for each sample were performed five times and the average values were used.
Adhesion measurements
The normal and shear adhesion of the adhesive samples were measured with custom-built equipment. The equipment consisted of motorized movable parts along the vertical and horizontal directions, load cells connected to the movable parts, and the sample holder. Adhesive samples with sizes of 1 ± 0.01 cm2 were attached to dry, moist, and submerged substrates (glass, Au, and porcine skin) with controlled preloads in the range of 20–300 kPa. After the application of the preload, the motorized part was moved along the vertical (normal adhesion test) or horizontal (shear adhesion test) axis with a retraction rate of 0.5 mm s−1 until the occurrence of separation at the adhesive/substrate interface. For the adhesion measurements against the moist substrate, a 3 μL water droplet was uniformly dispensed on the substrate to an area of 1 × 1 cm2. Subsequently, preloads were applied to the samples for a few seconds before the measurement. For the underwater adhesion measurements, the substrates were affixed to a mount in a small water bath placed on the adhesion test equipment. The adhesive samples were then submerged in the water bath for 10 min, followed by the pull-off and shear strength measurements. For each experimental condition, two specimens were tested and ten repeated adhesion measurements were conducted for each specimen. The averaged values and standard deviations of the measurements were used for the adhesion data.
Results and discussion
Fabrication and structure of the hydrogel adhesive
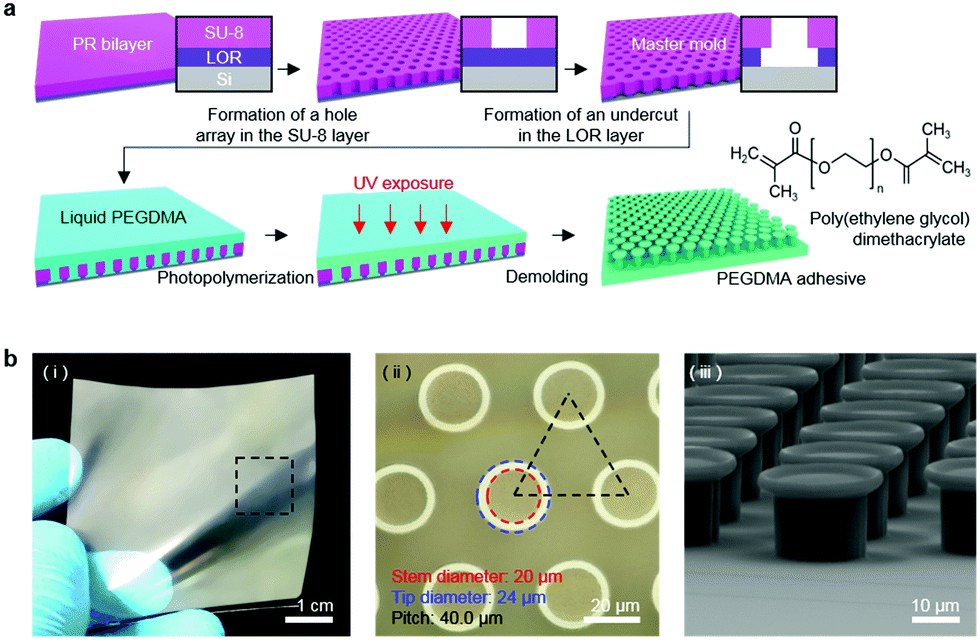
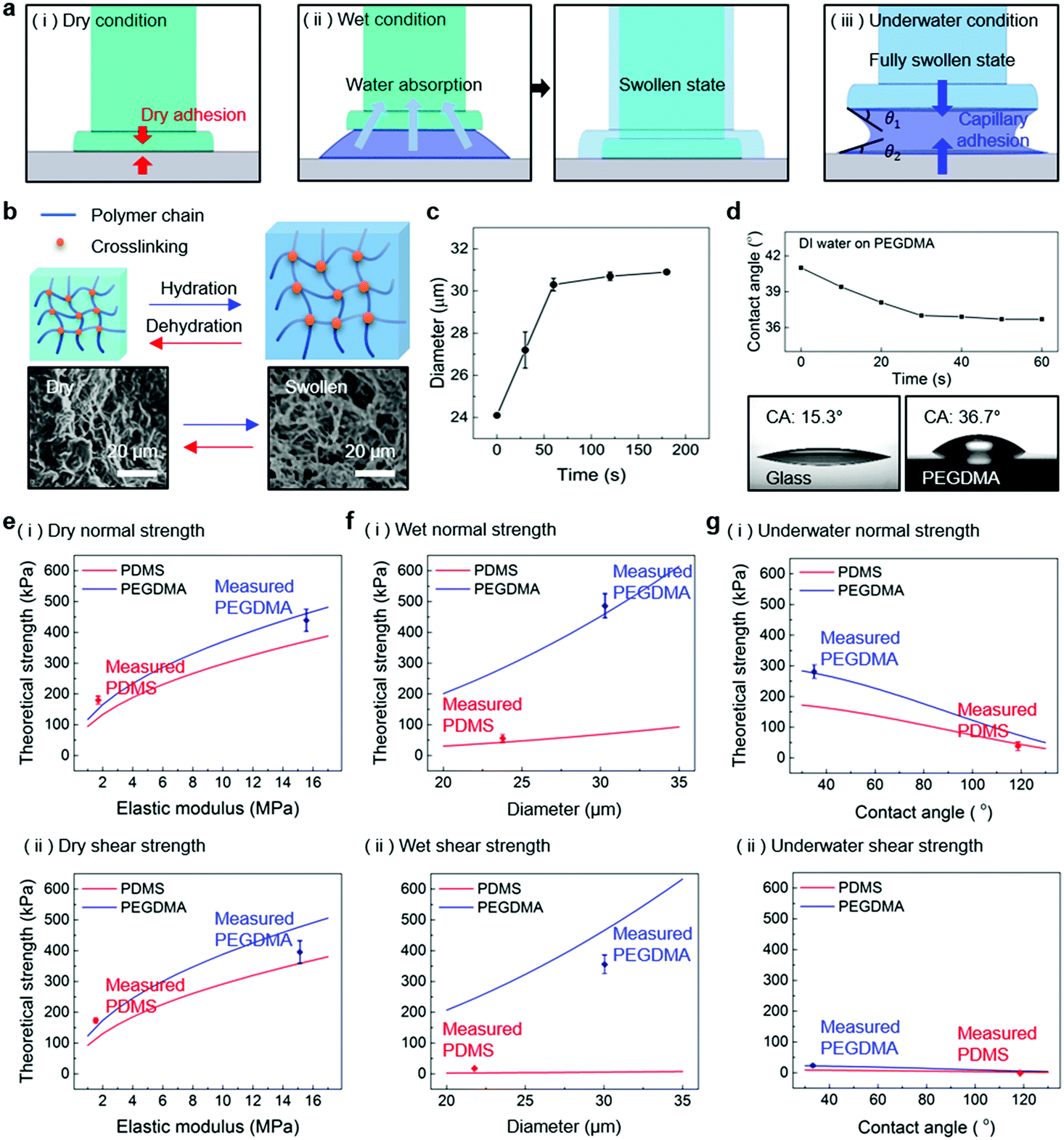
Fig. 1a shows a schematic of the hydrogel adhesive fabrication. As described above, the hydrogel adhesive was prepared using a replica molding technique against a patterned master using PEGDMA as a base material. PEGDMA is a hydrophilic polymeric hydrogel, which can absorb a large quantity of water. It can be rapidly cured by UV light irradiation within a few tens of seconds, which enables precise and scalable fabrication of diverse micro/nanostructures. In addition, its mechanical properties can be easily modulated by adjusting its concentration in water. Furthermore, it is biocompatible and thus can be utilized for various biomedical applications.3,19Fig. 1b shows the fabricated PEGDMA hydrogel adhesive. The resulting PEGDMA adhesive is in the form of a highly flexible film on which a uniform microstructure array is regularly formed (Fig. 1b-i). The individual microstructures have protruding heads on cylindrical stems (Fig. 1b-ii and iii). The protruding heads of the microstructures are beneficial to obtain high adhesion strengths against various surfaces based on van der Waals interactions without any surface treatment with adhesive chemical moieties. According to previous studies, the adhesion strengths of the microstructures with protruding heads are 5–26 times higher than those of simple microstructures with flat or hemispherical ends.34 The fabricated microstructures had a tip diameter of 24 μm, stem diameter of 20 μm, height of 25 μm, and spacing of 20 μm. It is worth noting that the fabricated PEGDMA in this study does not have a supporting layer (e.g., polyethylene terephthalate), as it leads to macroscopic bending of the adhesive film in the presence of water due to a mismatch in volume expansion between the swollen PEGDMA layer and nonswellable supporting layer.19
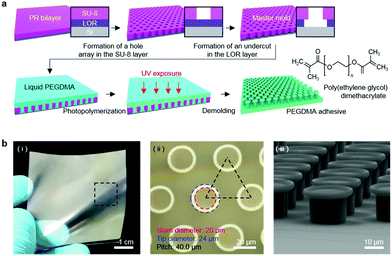 |
| | Fig. 1 (a) Schematic of the fabrication procedure of the PEGDMA adhesive. (b) (i) Photograph, (ii) microscopy image, and (iii) SEM image of the fabricated PEGDMA adhesive. | |
Wet adhesion behavior of the hydrogel adhesive
In order to evaluate the adhesion properties of the PEGDMA adhesive, we quantitatively measured the pull-off and shear strengths of the adhesive against a flat glass substrate under dry, wet, and underwater conditions (Fig. 2a). Under the wet conditions, the adhesion measurements were performed against a glass surface moistened with water. Under the underwater conditions, the adhesion measurements were performed against a submerged substrate. Adhesion of a polydimethylsiloxane (PDMS) adhesive was also evaluated for comparison. Fig. 2b-i and ii show the measured pull-off and shear strengths of the PDMS and PEGDMA adhesives with a preload of 20 kPa for the three different conditions. Overall, under the dry conditions, the PDMS and PEGDMA adhesives exhibited similar adhesion at a preload of 20 kPa. The normal adhesion strengths of the PDMS and PEGDMA adhesives were 152.3 and 160.6 kPa, respectively (Fig. 2c-i). The shear strengths of the PDMS and PEGDMA adhesives were 143.8 and 153.2 kPa, respectively (Fig. 2c-ii). However, for the moist substrate, the PDMS and PEGDMA adhesives exhibited completely different adhesion behaviors. The pull-off (normal) and shear strengths of the PDMS adhesive were significantly reduced to 40.2 and 24.2 kPa, respectively, which are 26.4% and 16.8%, respectively, of those against the dry substrate. This indicates that the PDMS adhesive has limited utility as a wet adhesive. On the contrary, the pull-off strength of the PEGDMA adhesive maintained a rather high value (143.4 kPa) against the wet substrate (Fig. 2c-i), which is equal to 89.3% of that for the dry substrate. In addition to the normal adhesion, the shear strength of the PEGDMA adhesive also remained at a high level (150 kPa) (Fig. 2c-ii), which is 97.9% of that for the dry substrate. The PEGDMA adhesive exhibited significant adhesion even to the submerged substrate (Fig. 2b and c). For the submerged substrate, the PEGDMA adhesive exhibited a normal adhesion strength of 120.2 kPa, whereas the PDMS adhesive exhibited a significantly degraded normal adhesion strength of 38.5 kPa. The shear adhesion of the PEGDMA adhesive decreased to 15.4 kPa for the submerged surface. However, this value is 2.75 times higher than that of the PDMS adhesive (5.6 kPa).
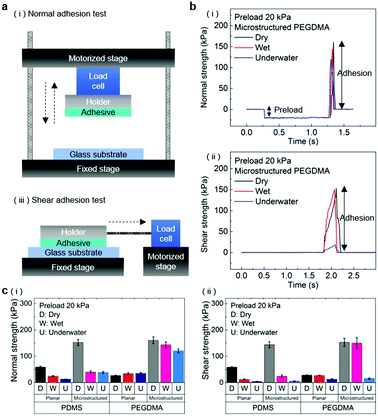 |
| | Fig. 2 (a) Experimental setups for (i) normal and (ii) shear adhesion measurements. (b) Representative examples of the measured (i) normal and (ii) shear adhesion under the three different conditions at a preload of 20 kPa. (c) (i) Normal and (ii) shear adhesion strengths of the PEGDMA and PDMS adhesives under the three different conditions at a preload of 20 kPa. Error bars represent standard deviations (N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) = 20). = 20). | |
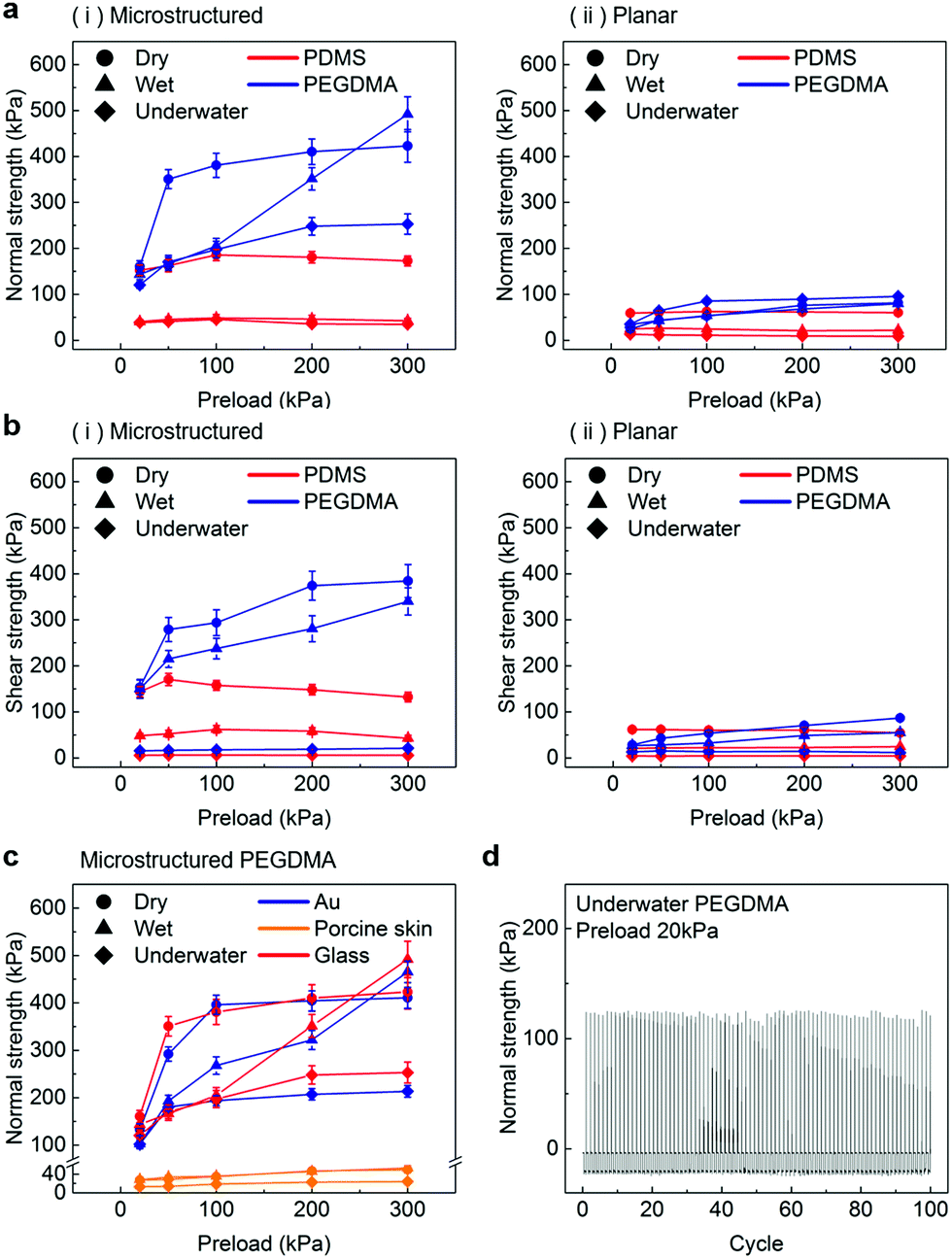
In order to further evaluate the wet adhesion performance of the PEGDMA adhesive, its normal and shear adhesion strengths were investigated at different preloads in the range of 20–300 kPa (Fig. 3). Under dry conditions (Fig. 4a-i), the maximum normal and shear adhesion strengths of the PEGDMA adhesive were 422.9 and 384.2 kPa at a preload of 300 kPa, respectively (Fig. 3a and b). In contrast, the maximum normal and shear adhesion strengths of the PDMS adhesive were 185.6 kPa (at a preload of 100 kPa) and 170.5 kPa (at a preload of 50 kPa), respectively. Theoretically, the normal and shear adhesion forces of microstructures with protruding tips are:64,65
| | 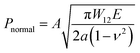 | (1a) |
| | 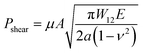 | (1b) |
where
A is the contact area,
W12 is the work of adhesion between the contacting surfaces,
E is the elastic modulus of the material used for the microstructures,
ν is the Poisson's ratio of the material,
a is the radius of the detached zone of the microstructures with protruding tips, and
μ is the friction coefficient. The higher surface energy (42.5 mJ m
−2) and elastic modulus (15.5 MPa) of PEGDMA than those of PDMS (surface energy: 20.5 mJ m
−2, elastic modulus: 1–2 MPa) lead to higher adhesion to the dry surface.
19,43,66,67 Our experimental adhesion-test results for the dry substrate are in agreement with the theoretical predictions (
μ ≈ 1.0, as shown in
Fig. 4e; see Fig. S1 (ESI
†) for the determination of the friction coefficient). It is worth noting that the relatively higher adhesion of the PEGDMA adhesive compared with that of the PDMS adhesive could be attributed to the hydrogen bonds between the PEG hydrogel and glass substrate.
68 To test this hypothesis, we performed additional adhesion measurements against Au and skin substrates (
Fig. 3c and Fig. S3, ESI
†). According to the measurement results, the PEGDMA adhesives exhibit overall similar adhesion strengths on the Au substrate compared to those on the glass substrate. It is observed that the adhesion strength of the planar PDMS on the glass was higher than that of the planar PEGDMA sample on the glass (
Fig. 2c). These results indicate that the hydrogen bonds between the PEGDMA adhesive and glass do not play a critical role in the measured macroscopic adhesion of the microstructured PEGDMA adhesive. The adhesion strengths of the PEGDMA adhesive on skin were much lower than those on the glass or Au substrates due to the relatively high surface roughness of skin compared with that of the flat glass or Au substrates.
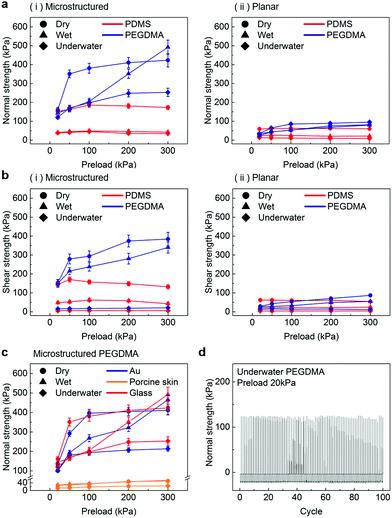 |
| | Fig. 3 (a) Normal and (b) shear adhesion strengths of the (i) microstructured and (ii) planar adhesives under the different conditions as a function of the preload. (c) Normal adhesion strengths of the microstructured PEGDMA adhesives on different substrates, namely glass, Au, and porcine skin. (d) Repeatability test results showing the normal adhesion strength of the PEGDMA adhesive under underwater conditions (preload: 20 kPa). Error bars represent standard deviations (for (a)–(c); N = 20). | |
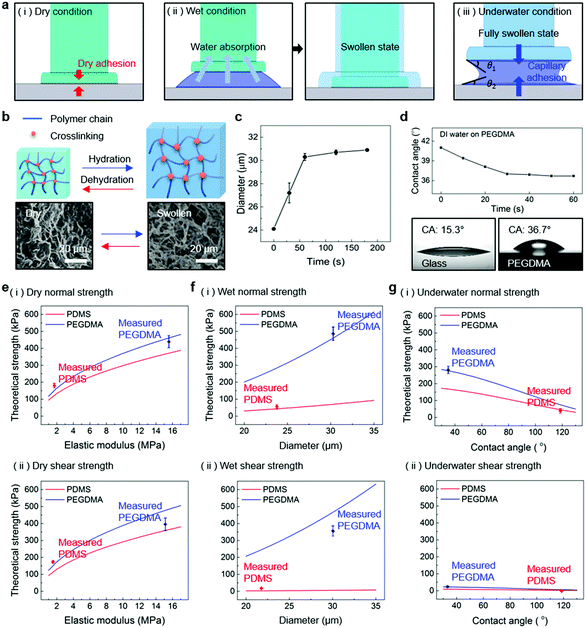 |
| | Fig. 4 (a) Conceptual illustrations showing the PEGDMA adhesive in contact with (i) dry, (ii) moist, and (iii) submerged substrates. (b) Conceptual illustration of the reversible swelling behavior of the PEGDMA hydrogel. (c) Diameter of the PEGDMA microstructures as a function of the water exposure time. (d) Water CA on the planar PEGDMA surface as a function of the time. Comparisons of the measured (i) normal and (ii) shear adhesion with the theoretical predictions for the (e) dry, (f) wet, and (g) underwater conditions. Error bars represent standard deviations (for (e)–(g); N = 20). | |
For a moist substrate, the PEGDMA adhesive exhibited maximum normal and shear strengths of 491.8 and 340 kPa at a preload of 300 kPa, respectively, which are similar to those of the PEGDMA adhesive for the dry substrate (Fig. 3a and b). On the contrary, for the moist substrate, the maximum normal and shear strengths of the PDMS adhesive were significantly reduced to 48.3 and 31.5 kPa (at a preload of 100 kPa), respectively (Fig. 3a and b). The remarkable adhesion strengths of the PEGDMA adhesive for the moist substrate mainly originated from the swelling behavior of the PEGDMA hydrogel, which can absorb a large amount of water. When the microstructured PEGDMA adhesive contacts the water on the substrate, the water is entrapped in the three-dimensional polymeric network of the PEGDMA microstructures (Fig. 4b), leading to partial or complete removal of the water on the substrate (Fig. 4a-ii). Accordingly, the PEGDMA adhesive can be in closer contact with the substrate. Furthermore, the diameter of the hydrogel microstructures increased owing to the swelling of the hydrogel (Fig. 4c). Consequently, a high level of adhesion can be obtained with the PEG-hydrogel-based adhesive for the moist substrate. The maximum experimental adhesion values for this case followed eqn (1) well (Fig. 4f). On the contrary, in the case of the PDMS adhesive, the water on the substrate could not be perfectly removed despite the hydrophobic nature of PDMS. Consequently, the water remaining on the substrate hinders close contact between the microstructure and substrate, leading to the remarkably reduced normal and shear adhesion strengths.1,6,8,18–20 In this case, the adhesion originated mainly from capillary adhesion. The capillary adhesion between two adjacent surfaces is expressed as:69,70
| | 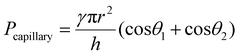 | (2) |
where
γ is the surface tension of water,
r is the contact radius,
h is the thickness of the water layer, and
θ1 and
θ2 are the water CAs at the two surfaces. The adhesion behavior of the PDMS adhesive for the wet surface agrees well with
eqn (2) (
Fig. 4g).
The PEGDMA adhesive exhibited significant adhesion even to the submerged surface. The maximum normal adhesion of the PEGDMA adhesive reached 253 kPa (at a preload of 300 kPa), whereas that of the PDMS adhesive was 42.2 kPa (at a preload of 100 kPa). The superior underwater adhesion of the PEGDMA adhesive is based on enhanced capillary adhesion owing to the water-friendly characteristic of the PEGDMA hydrogel. According to eqn (2), the capillarity-induced normal adhesion strength is higher for a hydrophilic material with a lower water CA. In the case of PEGDMA, the water CA monotonously decreased with the increase in the water exposure time, reaching 36° at equilibrium (Fig. 4d), while that of PDMS was approximately 120° regardless of the water exposure time. The theoretically calculated adhesion strength of the PEGDMA adhesive is 274.8 kPa for a submerged glass substrate, while that of the PDMS adhesive is 43.7 kPa, which agrees well with our experimental observations (Fig. 4g). The maximum shear adhesion strength of the PEGDMA adhesive under the underwater conditions was 21.2 kPa, significantly lower than the maximum normal adhesion value. The reduced shear strength for the underwater surface can be explained as stable friction cannot be established under the wet conditions owing to the water layer, which obstructs the direct solid–solid contact. The friction coefficients of water-lubricated PDMS and PEGDMA were estimated to be ∼0.05 and 0.08, respectively (Fig. S1, ESI†). Accordingly, the underwater shear strength (Pshear = μPcapillary) is significantly reduced compared to the underwater normal strength. Nevertheless, the underwater shear strength of the PEGDMA adhesive (21.2 kPa) is 3.4 times higher than that of the PDMS adhesive (6.24 kPa). Furthermore, the PEGDMA adhesion was highly reversible. The high level of underwater adhesion could be maintained over repeated cycles of adhesion tests (Fig. 3d). For all the experimental conditions, the PEGDMA and PDMS adhesives with bioinspired microstructures exhibited significantly higher adhesion strengths than those of nonpatterned planar PEGDMA and PDMS samples (Fig. 3a-ii and b-ii). This indicates that the bioinspired microstructures of our adhesive have proper structural topographies as robust and reversible adhesives. Our adhesive is also highly flexible and is completely made of biocompatible PEG hydrogel. Therefore, it holds strong potential for applications in flexible and reversible skin-and tissue-attachable patches. Indeed, although the adhesion strength of the PEGDMA adhesives was lower on skin as compared with that on the flat glass or Au substrates, the skin adhesion of PEGDMA was in the range of ∼25–45 kPa, which is comparable to those of existing skin adhesives.17,71 Therefore, by incorporating emerging flexible electronics and functional chemical moieties for more stable skin and tissue adhesion, this PEGDMA adhesive can be used in flexible patch-type platforms for advanced biomedical or healthcare devices.
Conclusions
We demonstrated the high and reversible wet and underwater adhesion performance of a bioinspired PEG hydrogel adhesive. By simply combining the water-absorbing and swelling properties of the polymeric hydrogel and bioinspired microscale architectures, high normal and shear adhesion strengths could be obtained with the adhesive for moist and submerged substrates, which surpassed those of previously reported bioinspired reversible wet adhesives. In addition to that to the wet and underwater surfaces, our hydrogel adhesive exhibited remarkable adhesion to a dry surface. Therefore, our hydrogel adhesive represents a universal adhesive that can strongly and reversibly adhere to dry, moist, and submerged surfaces. This bioinspired hydrogel adhesive with smart adhesion performance could be used as a versatile and advanced adhesive for various biomedical applications that require stable and repeatable adhesion under diverse dry and wet environments.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Institute for Information & communications Technology Promotion (IITP) grant funded by the Korea government (MSIP) (No. 2017000910001100, 20180007560011001).
References
- Y. H. Zhao, Y. Wu, L. Wang, M. M. Zhang, X. Chen, M. J. Liu, J. Fan, J. Q. Liu, F. Zhou and Z. K. Wang, Nat. Commun., 2017, 8, 2218 CrossRef PubMed.
- D. K. Wainwright, T. Kleinteich, A. Kleinteich, S. N. Gorb and A. P. Summers, Biol. Lett., 2013, 9, 20130234 CrossRef PubMed.
- H. H. Park, M. Seong, K. Sun, H. Ko, S. M. Kim and H. E. Jeong, ACS Macro Lett., 2017, 6, 1325–1330 CrossRef CAS.
- Y. F. Ma, S. H. Ma, Y. Wu, X. W. Pei, S. N. Gorb, Z. K. Wang, W. M. Liu and F. Zhou, Adv. Mater., 2018, 30, 1801595 CrossRef PubMed.
- J. Liu, Q. L. Yang, J. J. Xu, K. S. Liu, L. Guo and L. Jiang, Prog. Chem., 2012, 24, 1946–1954 CrossRef CAS.
- M. Li, J. Xie, Q. W. Dai, W. Huang and X. L. Wang, J. Mech. Behav. Biomed. Mater., 2018, 78, 266–272 CrossRef CAS PubMed.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS PubMed.
- H. Ko, M. Seong and H. E. Jeong, Soft Matter, 2017, 13, 8419–8425 RSC.
- K. Autumn, C. Majidi, R. E. Groff, A. Dittmore and R. Fearing, J. Exp. Biol., 2006, 209, 3558–3568 CrossRef CAS PubMed.
- G. Carbone and E. Pierro, Small, 2012, 8, 1449–1454 CrossRef CAS PubMed.
- H. E. Jeong, J. K. Lee, H. N. Kim, S. H. Moon and K. Y. Suh, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5639–5644 CrossRef CAS PubMed.
- J. Jeong, J. Kim, K. Song, K. Autumn and J. Lee, J. R. Soc., Interface, 2014, 11, 140627 CrossRef PubMed.
- S. M. Kang, Int. J. Precis. Eng. Manuf. – Green Technol., 2016, 3, 189–192 CrossRef.
- M. Rohrig, M. Thiel, M. Worgull and H. Holscher, Small, 2012, 8, 3009–3015 CrossRef PubMed.
- D. P. Wang, A. W. Zhao, R. Jiang, D. Li, M. F. Zhang, Z. B. Gan, W. Y. Tao, H. Y. Guo and T. Mei, Appl. Surf. Sci., 2012, 259, 93–98 CrossRef CAS.
- M. Zhou, N. Pesika, H. B. Zeng, Y. Tian and J. Israelachvili, Friction, 2013, 1, 114–129 CrossRef CAS.
- I. Hwang, H. N. Kim, M. Seong, S. H. Lee, M. Kang, H. Yi, W. G. Bae, M. K. Kwak and H. E. Jeong, Adv. Healthcare Mater., 2018, 7, 1800275 CrossRef PubMed.
- L. Heepe, A. E. Kovalev and S. N. Gorb, Beilstein J. Nanotechnol., 2014, 5, 903–909 CrossRef CAS PubMed.
- H. Yi, M. Seong, K. Sun, I. Hwang, K. Lee, C. Cha, T. I. Kim and H. E. Jeong, Adv. Funct. Mater., 2018, 28, 1706498 CrossRef.
- A. Y. Stark, I. Badge, N. A. Wucinich, T. W. Sullivan, P. H. Niewiarowski and A. Dhinojwala, Proc. Natl. Acad. Sci. U.
S. A., 2013, 110, 6340–6345 CrossRef CAS PubMed.
- J. W. Yang, R. B. Bai and Z. G. Suo, Adv. Mater., 2018, 30, 1800671 CrossRef PubMed.
- J. Li, A. D. Celiz, J. Yang, Q. Yang, I. Wamala, W. Whyte, B. R. Seo, N. V. Vasilyev, J. J. Vlassak, Z. Suo and D. J. Mooney, Science, 2017, 357, 378–381 CrossRef CAS PubMed.
- B. K. Ahn, J. Am. Chem. Soc., 2017, 139, 10166–10171 CrossRef CAS PubMed.
- Y. Xu, Q. H. Liu, A. Narayanan, D. Jain, A. Dhinojwala and A. Joy, Adv. Mater. Interfaces, 2017, 4, 1700506 CrossRef.
- M. A. North, C. A. Del Grosso and J. J. Wilker, ACS Appl. Mater. Interfaces, 2017, 9, 7866–7872 CrossRef CAS PubMed.
- B. P. Lee, C. Y. Chao, F. N. Nunalee, E. Motan, K. R. Shull and P. B. Messersmith, Macromolecules, 2006, 39, 1740–1748 CrossRef CAS.
- S. K. Clancy, A. Sodano, D. J. Cunningham, S. S. Huang, P. J. Zalicki, S. Shin and B. K. Ahn, Biomacromolecules, 2016, 17, 1869–1874 CrossRef CAS PubMed.
- H. Shao and R. J. Stewart, Adv. Mater., 2010, 22, 729–733 CrossRef CAS PubMed.
- Y. C. Choi, J. S. Choi, Y. J. Jung and Y. W. Cho, J. Mater. Chem. B, 2014, 2, 201–209 RSC.
- B. Soltannia and D. Sameoto, ACS Appl. Mater. Interfaces, 2014, 6, 21995–22003 CrossRef CAS PubMed.
- H. Jiang, E. W. Hawkes, C. Fuller, M. A. Estrada, S. A. Suresh, N. Abcouwer, A. K. Han, S. Q. Wang, C. J. Ploch, A. Parness and M. R. Cutkosky, Sci. Robot., 2017, 2, 4545 CrossRef.
- H. Kasem, A. Tsipenyuk and M. Varenberg, Soft Matter, 2015, 11, 2909–2915 RSC.
- M. K. Kwak, H. E. Jeong, W. G. Bae, H. S. Jung and K. Y. Suh, Small, 2011, 7, 2296–2300 CrossRef CAS PubMed.
- H. Shahsavan, S. M. Salili, A. Jakli and B. X. Zhao, Adv. Mater., 2017, 29, 1604021 CrossRef PubMed.
- Y. Wang, H. Hu, J. Y. Shao and Y. C. Ding, ACS Appl. Mater. Interfaces, 2014, 6, 2213–2218 CrossRef CAS PubMed.
- W. Federle, J. Exp. Biol., 2006, 209, 2611–2621 CrossRef PubMed.
- C. Greiner, E. Arzt and A. del Campo, Adv. Mater., 2009, 21, 0801548 CrossRef.
- G. Huber, H. Mantz, R. Spolenak, K. Mecke, K. Jacobs, S. N. Gorb and E. Arzt, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16293–16296 CrossRef CAS PubMed.
- S. Kim, M. Sitti, T. Xie and X. C. Xia, Soft Matter, 2009, 5, 3689–3693 RSC.
- H. Ko, H. Yi and H. E. Jeong, Int. J. Precis. Eng. Manuf. – Green Technol., 2017, 4, 273–280 CrossRef.
- P. C. Lin, S. Vajpayee, A. Jagota, C. Y. Hui and S. Yang, Soft Matter, 2008, 4, 1830–1835 RSC.
- J. Purtov, M. Frensemeier and E. Kroner, ACS Appl. Mater. Interfaces, 2015, 7, 24127–24135 CrossRef CAS PubMed.
- H. Yi, M. Kang, M. K. Kwak and H. E. Jeong, ACS Appl. Mater. Interfaces, 2016, 8, 22671–22678 CrossRef CAS PubMed.
- D. Sameoto, H. Sharif and C. Menon, J. Adhes. Sci. Technol., 2012, 26, 2641–2652 CrossRef CAS.
- M. P. Murphy, B. Aksak and M. Sitti, Small, 2009, 5, 170–175 CrossRef CAS PubMed.
- Y. Menguc, S. Y. Yang, S. Kim, J. A. Rogers and M. Sitti, Adv. Funct. Mater., 2012, 22, 1246–1254 CrossRef.
- S. H. Ma, D. A. Wang, Y. M. Liang, B. Q. Sun, S. N. Gorb and F. Zhou, Small, 2015, 11, 1131–1137 CrossRef CAS PubMed.
- N. W. Rizzo, K. H. Gardner, D. J. Walls, N. M. Keiper-Hrynko, T. S. Ganzke and D. L. Hallahan, J. R. Soc., Interface, 2006, 3, 441–451 CrossRef CAS PubMed.
- H. Yi, I. Hwang, M. Sung, D. Lee, J. H. Kim, S. M. Kang, W. G. Bae and H. E. Jeong, Int. J. Precis. Eng. Manuf. – Green Technol., 2014, 1, 347–351 CrossRef.
- A. P. Defante, A. Nyarko, S. Kaur, T. N. Burai and A. Dhinojwala, Langmuir, 2018, 34, 4084–4094 CrossRef CAS PubMed.
- M. K. Chaudhury and G. M. Whitesides, Langmuir, 1991, 7, 1013–1025 CrossRef CAS.
- P. Rao, T. L. Sun, L. Chen, R. Takahashi, G. Shinohara, H. Guo, D. R. King, T. Kurokawa and J. P. Gong, Adv. Mater., 2018, 30, 1801884 CrossRef PubMed.
- R. Kempaiah and Z. H. Nie, J. Mater. Chem. B, 2014, 2, 2357–2368 RSC.
- K. Y. Seong, M. S. Seo, D. Y. Hwang, E. D. O'Cearbhaill, S. Sreenan, J. M. Karp and S. Y. Yang, J. Controlled Release, 2017, 265, 48–56 CrossRef CAS PubMed.
- S. Y. Yang, E. D. O’Cearbhaill, G. C. Sisk, K. M. Park, W. K. Cho, M. Villiger, B. E. Bouma, B. Pomahac and J. M. Karp, Nat. Commun., 2013, 4, 1702 CrossRef PubMed.
- H. Yuk, T. Zhang, S. T. Lin, G. A. Parada and X. H. Zhao, Nat. Mater., 2016, 15, 190–198 CrossRef CAS PubMed.
- S. H. Ma, M. Scaraggi, P. Lin, B. Yu, D. A. Wang, D. Dini and F. Zhou, J. Phys. Chem. C, 2017, 121, 8452–8463 CrossRef CAS.
- H. C. Gu, B. F. Ye, H. B. Ding, C. H. Liu, Y. J. Zhao and Z. Z. Gu, J. Mater. Chem. C, 2015, 3, 6607–6612 RSC.
- D. J. Guo, H. Zhang, J. B. Li, S. M. Fang, Z. D. Dai and W. Tan, J. Mater. Chem. B, 2013, 1, 379–386 RSC.
- C. Heo, C. Jeong, H. S. Im, J. U. Kim, J. Woo, J. Y. Lee, B. Park, M. Suh and T. I. Kim, Nanoscale, 2017, 9, 17743–17751 RSC.
- J. H. Lee, Y. M. Lee, Y. H. Kim, J. Y. Lee, S. J. Choi and P. J. Yoo, Small, 2011, 7, 2587–2592 CrossRef CAS PubMed.
- M. K. Kwak, H. E. Jeong, T. I. Kim, H. Yoon and K. Y. Suh, Soft Matter, 2010, 6, 1849–1857 RSC.
- S. H. Lee, S. W. Kim, C. W. Park, H. E. Jeong, J. G. Ok and M. K. Kwak, Int. J. Precis. Eng. Manuf. – Green Technol., 2017, 4, 177–181 CrossRef.
- G. Carbone, E. Pierro and S. N. Gorb, Soft Matter, 2011, 7, 5545–5552 RSC.
- Y. Tian, N. Pesika, H. B. Zeng, K. Rosenberg, B. X. Zhao, P. McGuiggan, K. Autumn and J. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19320–19325 CrossRef CAS PubMed.
- E. S. Pane, J. E. A. Palamara and H. H. Messer, Dent. Mater., 2012, 28, E150–E159 CrossRef CAS PubMed.
- H. Seong, K. Pak, M. Joo, J. Choi and S. G. Im, Adv. Electron. Mater., 2016, 2, 1500209 CrossRef.
- A. H. Hofman, I. A. van Hees, J. Yang and M. Kamperman, Adv. Mater., 2018, 30, 1704640 CrossRef PubMed.
- E. Cheung and M. Sitti, J. Adhes. Sci. Technol., 2008, 22, 569–589 CrossRef CAS.
- H. Peisker, L. Heepe, A. E. Kovalev and S. N. Gorb, J. R. Soc., Interface, 2014, 11, 140752 CrossRef PubMed.
- S. Baik, D. W. Kim, Y. Park, T. J. Lee, S. H. Bhang and C. Pang, Nature, 2017, 546, 396–400 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb02598c |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Hoon Eui
Jeong
b and
Hoon Eui
Jeong