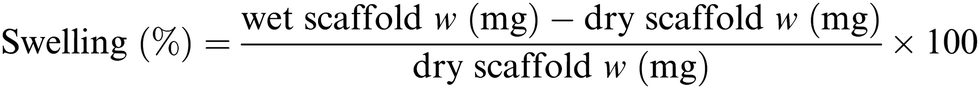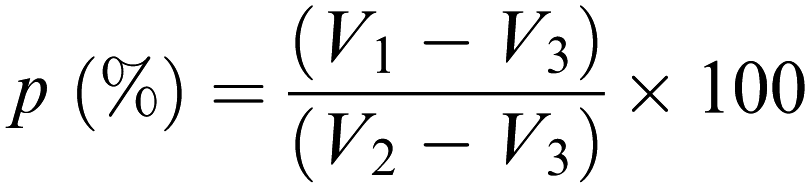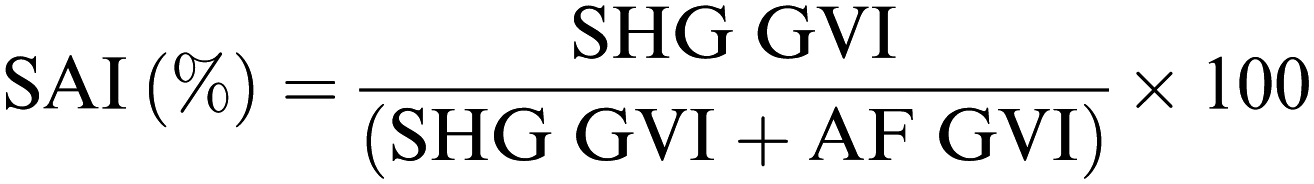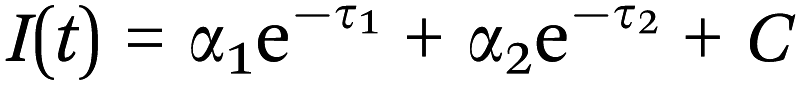Non-invasive characterization of hybrid gelatin:poly-L-lactide electrospun scaffolds using second harmonic generation and multiphoton imaging†
Received
2nd August 2018
, Accepted 29th August 2018
First published on 18th September 2018
Abstract
Hybrid scaffolds composed of synthetic polymers and naturally occurring components have become more relevant in the field of tissue engineering and regenerative medicine. Synthetic polymers are responsible for scaffold durability, strength and structural integrity; however, often do not provide biological signals. Introducing a biological component leads to more advanced and biocompatible scaffolds. In order to use these scaffolds as implants, a deeper knowledge of material characteristics and the impact of the biological component on the scaffold mechanical properties are required. Furthermore, it is necessary to implement fast, easy and non-invasive methods to determine material characteristics. In this work, we aimed to generate gelatin-poly-L-lactide (PLA) hybrids via electrospinning with defined, controllable and tunable scaffold characteristics. Using Raman microspectroscopy, we demonstrated the effectiveness of the cross-linking reaction and evaluated the increasing PLA content in the hybrid scaffolds with a non-invasive approach. Using multiphoton microscopy, we showed that gelatin fibers electrospun from a fluorinated solvent exhibit a second harmonic generation (SHG) signal typical for collagen-like structures. Compared to pure gelatin, where the SHG signal vanishes after cross-linking, the signal could be preserved in the hybrid scaffolds even after cross-linking. Furthermore, we non-invasively imaged cellular growth of human dermal fibroblasts on the hybrid electrospun scaffolds and performed fluorescence lifetime imaging microscopy on the cell-seeded hybrids, where we were able to discriminate between cells and scaffolds. Here, we successfully employed non-invasive methods to evaluate scaffold characteristics and investigate cell–material interactions.
1. Introduction
The extracellular matrix (ECM) is a three-dimensional (3D), complex fibrous network consisting of various proteins, such as glycoproteins, proteoglycans, collagens, elastic fibers, as well as growth factors, cytokines and other important small molecules.1,2 The ECM serves as a scaffold for cells and additionally provides biochemical signals, which impact cellular behavior.3–6 Blend nanofibrous electrospun scaffolds represent an emerging class of nanostructures that can mimic the ECM of native tissues, and thus help to support damaged organs and favor their regeneration.7–10 Hybrid scaffolds with unique mechanical, biochemical and structural properties can be generated by combining two or more polymers.11 Blend electrospun scaffolds have already been applied in the field of tissue engineering and as drug delivery systems.9,12 Many scientists have demonstrated that nanofibrous scaffolds are capable of mimicking and reorganizing the ECM to sustain damaged or pathological areas.13 In addition, it has been shown that they favor tissue regeneration and induce healing processes.14–17 The electrospinning process leads to 3D structures with a high surface to volume ratio, composed of nanofibers and pores with variable sizes.18–22 Electrospun scaffolds exhibit strong mechanical properties while maintaining a very low density.23,24 Moreover, their degradation profile can be predicted before being implanted in the human body or even modified later in situ.25,27 Thus, the implanted scaffolds slowly break down into non-toxic fragments in a predictable manner, while the native tissue gradually grows and replaces the scaffold.25,26 To date, many different synthetic and natural or naturally derived polymers have been electrospun into nanofibrous scaffolds.27,28 Among them, the most widely electrospun synthetic polymers are linear aliphatic polyesters like polylactic acid,29–31 polyglycolic acid (PGA)32 and their copolymer, poly(lactic-co-glycolic) acid (PLGA).33–35 All of these materials are biocompatible, can be easily obtained from cost-effective raw material sources,29–35 and have already been widely used in soft tissue regeneration.36,37 Synthetic materials are strong, cheap and reliable but they unfortunately share no biochemical signatures. This aspect is desired in some cases but in others a specific interaction with the target tissue is required. To obtain biomimetic nanofibers, an appealing approach has been the direct electrospinning of natural or naturally derived polymers.38–40 To date, collagens, gelatin, fibrinogen, chitosan and alginate have all been used as polymers to obtain nanofibrous structures by electrospinning.40 Many of these macromolecules retain cell-binding sites and biomolecular signatures that can favor cell–material interactions.41 The harvesting and processing of natural polymers, however, is not as straightforward as for synthetic polymers, since their unique and specific properties are strongly influenced by temperature and solvent interaction.41,42 Thus, a greater attention should be paid when working with these polymers in order to not alter or denature them.42 In the last years, there is a growing interest in hybrid scaffolds, since they have certain advantages compared to single-component systems.7–12 By blending two or more polymers the resulting scaffolds combine the characteristics of the different polymers or even exhibit new unique features.7–11 While each single polymer unlikely shares the chemical composition and the structural properties of the native tissue, polymers used in combination can better recapitulate the complexity of the ECM. Starting from blend solutions also enables the electrospinning of polymers that are challenging to process alone.43–45 However, the development of well-blended hybrid scaffolds by electrospinning is difficult due to the poor miscibility of the different polymers.46 Insufficiently blended polymeric nanofibers exhibit weak mechanical strength and have unpredictable material properties as a result of inhomogeneity.47 Thus, the production of hybrid electrospun nanostructures, which benefit from the favorable biological properties of the natural polymers and from the mechanical properties of the synthetic polymers still remains a great challenge in tissue engineering and regenerative medicine. In this work, we present the development and characterization of different hybrid electrospun scaffolds composed of blended gelatin (GE) and PLA, a natural and a synthetic polymer, respectively. GE consists of a mixture of water-soluble protein fragments, with the same amino acid composition as collagen from which it is obtained through partial hydrolysis.48 GE has been successfully employed in the production of biomaterials profiting from its biodegradability, biocompatibility and commercial availability at low cost.49 GE has advantages over collagen, such as lower immunogenicity50–53 and better solubility in aqueous systems.52 Unmodified GE increases cell adherence and proliferation, but has unfortunately low mechanical resistance.53 Due to its high water solubility, which represents an advantage for processing, cross-linking is mandatory for further application in biomedicine. Even though various harmless, clean methodologies are available, cross-linking potentially alters material properties and biocompatibility.54–56 GE can be obtained from different animal collagens.57–60 Mammalian GE is rich in domains able to bind to cell–surface receptors and to other ECM proteins, such as fibronectin, thus offering an excellent substrate for attachment and proliferation of adherent cells.61 In addition, GE undergoes collagenase-mediated hydrolysis, which allows biologically driven remodeling of GE-based scaffolds in vivo,62 and material resorption without toxic residuals.63 Cell adhesion on PLA is instead low,64 but its mechanical resistance and elasticity are higher compared to GE.65,66 PLA is a widely used aliphatic polyester,67 which can be chemically synthesized either by poly-condensation of lactic acid or by ring-opening polymerization of lactide, a cyclic dimer of lactic acid.68 One attractive feature of PLA is that it is resorbed by the human body following hydrolytic and enzymatic digestion.69In vivo, PLA hydrolysis occurs by the cleavage of its ester bonds resulting in non-toxic lactic acid, which is finally eliminated as CO2 and H2O via the Krebs cycle.70,71 Amorphous poly-lactide is eliminated within a year,72 while isotactic PLA full bio-resorption requires a longer time.73,74 Thus, PLA is the polymer of choice for the preparation of biomaterials when a longer exploitation time is required.
In this work, we aimed to produce and characterize hybrid GE:PLA scaffolds with controlled and tunable porosity, wettability and structural properties in order to be able to produce well-defined ECM mimicking hybrid structures.
2. Materials and methods
2.1. Electrospinning
PLA (Sigma-Aldrich #93578; Mn 59000, Mw 101 kDa) and GE (gelatin from bovine skin, type B, cell culture tested, Mw 50–100 kDa, Bloom strength ∼225 Bloom) were purchased from Sigma-Aldrich (Steinheim, Germany). After polymer addition the solutions were incubated at 37 °C under permanent magnetic stirring for 24 hours. 1,1,1,3,3,3-Hexafluoro-2-propanol (HFP, Sigma-Aldrich) was used as solvent for all experiments. All electrospinning experiments were performed with a customized electrospinning device.75 The parameters used for the electrospinning experiments are reported in Table 1. In all cases, an 18G stainless steel needle was employed at a distance of 19 cm from the collector. The applied voltage was 19.5 kV. The used collector was round with a diameter of 9 cm.
Table 1 Parameters used for electrospinning
| Polymer(s) |
Final w/v% |
Final volume (mL) |
Flow-rate (mL h−1) |
| GE |
10 |
3 |
1.2 |
GE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PLA = 4:1 :PLA = 4:1 |
12.5 |
3 |
1.6 |
| GE:PLA = 5:2 |
14 |
3 |
1.6 |
| GE:PLA = 1:1 |
15 |
3 |
2.1 |
2.2. Cross-linking procedure
All solvents and reagents were purchased from Sigma-Aldrich unless stated otherwise. For cross-linking, a solution of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl, Novabiochem®, EMD Merck Millipore, Darmstadt, Germany) and N-hydroxysuccinimide (NHS) in equimolar ratio, both at the concentration of 50 mM, was used. EtOH 90%/ddH2O was used as solvent. After immersion, the scaffolds were shaken in the cross-linking mixture at room temperature (r.t.) for 24 hours using an orbital shaker (60 rpm). The scaffolds were then gently dried on filter paper and washed with ddH2O for another 24 hours. After the washing procedure, the excess of water was removed using a filter paper. The scaffolds were finally immersed in 70% EtOH/ddH2O for 30 minutes, then dried on filter paper and allowed to stay overnight at r.t. to complete evaporate any solvent residual.
2.3. Scanning electron microscopy (SEM)
The morphology of the electrospun scaffolds was determined using a scanning electron microscope (1530 VP, Zeiss, Jena, Germany). After platinum sputter coating, images were acquired at a distance of 8 mm from the detector, a voltage of 15 kV and different magnifications. The ImageJ® software supplied with the DiameterJ plug-in was used for fiber diameter and pores size analyses.
2.4. Raman microspectroscopy
The inverse Raman system and acquisition parameters used in this study were previously described.76 Briefly, a 784 nm diode laser with an output laser power of 85 mW was focused through a water-immersion objective (60×, NA 1.2, Olympus, Tokyo, Japan). The total acquisition time per spectrum was 100 s (10 × 10 s acquisitions). All Raman spectra were analyzed in the range from 400 to 1800 cm−1. Spectra were background-subtracted, baseline-corrected using OPUS (Bruker Optics, Ettlingen, Germany) and vector-normalized using Unscrambler X 10.3 (Camo, Oslo, Norway) as previously described.76 Principal component analysis (PCA) was performed using Unscrambler X 10.3 to reduce spectral variables and identify spectral differences among the compared samples. Seven PCs were calculated for each PCA. PC loadings were considered in detail to identify the molecular components that were relevant for the comparison of the spectra.
2.5. Swelling test analysis
For each sample 16 different pieces obtained from four different scaffolds were employed. Each scaffold piece was immersed in 10 mL ddH2O for 1 hour at r.t. using an orbital shaker (60 rpm). In case water drops on the scaffold surface were clearly visible, the excess water was gently removed using filter paper. The rate of water absorbed by each piece was calculated according to following formula (Scheme 1):77
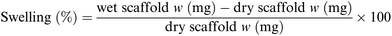 |
| | Scheme 1 Formula used for the swelling test.77 | |
2.6. Liquid displacement method
The porosity of the scaffolds was estimated using the liquid displacement method, as previously described.77–79 Absolute ethanol was used as the displacement liquid. Scaffold samples were immersed in a cylinder containing a known volume of absolute ethanol (V1) for 30 minutes. After that time, the scaffold was gently pressed to remove air bubbles and the final volume represented by ethanol and ethanol-impregnated scaffold was recorded as V2. Finally, the ethanol-impregnated scaffold was removed and the residual ethanol volume in the cylinder was recorded as V3. The porosity was calculated as follows (Scheme 2):78
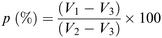 |
| | Scheme 2 Formula used for the porosity measurement according to the liquid displacement method.78 | |
2.7. Contact angle measurements
Hydrophilicity of the electrospun substrates was analyzed using contact angle measurements with an OCA 40 device (DataPhysics Instruments GmbH, Filderstadt, Germany). A water drop with the volume of 2 μL was placed onto the sample and the contact angle was measured 10 seconds after water deposition using a video setup and the SCA20 software (DataPhysics Instruments) as previously described.80 Final results were calculated from 16 measurements obtained from 4 different scaffold pieces for each sample.
2.8. Uniaxial tensile testing
Electrospun scaffolds were cut into 10 mm × 20 mm rectangular pieces and clamped into the uniaxial tensile testing device (Electroforce 5500, ElectroForce® Systems Group, Bose Corporation, Minnesota, USA). The exact sample dimensions were determined before each measurement and recorded with the software for further calculations of the Young's modulus and the tensile strength. The scaffolds were pulled to failure by applying a stretch of 0.025 mm s−1. The Young's modulus was calculated from the initial linear slope of the stress versus strain curve for each measurement. Measured values are presented as average ± standard deviation from 16 measurements obtained from 4 different scaffold pieces for each sample.
2.9. Cell culture and seeding
All research was carried out in compliance with the rules for investigation of human subjects, as defined in the Declaration of Helsinki. This study was carried out in accordance with the institutional guidelines and was approved by the local research Ethics Committee (F-2012-078). Human dermal fibroblasts (HDFs) were isolated by enzymatic digestion as previously described.81 Cells were cultured in Dulbecco's modified eagle medium (DMEM, with L-glutamine, Gibco™, Life Technologies GmbH, Darmstadt, Germany) supplemented with 10% fetal calf serum (FCS, PAA Laboratories, Pasching, Austria) and 1% penicillin/streptomycin (100 U mL−1 penicillium and 100 μg mL−1 streptomycin, Life Technologies GmbH). Cells were cultured in an incubator at 37°C and in a 5% CO2 atmosphere. Cell culture medium was changed every 3 days and cells were passaged or seeded using trypsin–EDTA (15090046, PAA Laboratories) at approximately 70% confluence. When seeding the cells directly on electrospun scaffolds, HDFs were seeded in 12-well plates using inserts (CellCrown™, Scaffdex, Tampere, Finland) in number of 2 × 105 per insert.
2.10.
In vitro cytotoxicity assay
According to an ISO 10993-5 accredited protocol, HDFs were exposed to an extract of the samples. The electrospun scaffolds were sterilized with 70% ethanol for 30 minutes. 6 cm2 of each sample was then incubated in 1 mL FCS- and antibiotic-free DMEM medium for 72 hours. Each extract was prepared in triplicate. HDFs seeded in 96-well plates (2000 cells per well) were then exposed for 24 hours to the extracts supplied with 10% FCS. The extraction medium was removed, the cells washed twice with phosphate buffer saline 1× (PBS, Gibco™ by Life Technologies GmbH) and a tetrazolium salt ([3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium], MTS) assay (CellTiter 96Aqueous One Solution Cell Proliferation Assay, Promega, Mannheim, Germany) was performed as per the manufacturer's protocol. Briefly, 20 μL of MTS solution was added to 100 μL of culture media. After 35 minutes incubation at 37 °C, the absorbance of each well was measured at 492 nm using a TECAN® (Crailsheim, Germany) Infinite 200 Reader. The test was performed for a blind, a negative control (NC; fresh DMEM + 10% v FCS) and a sodium dodecyl sulfate (SDS, Life Technologies GmbH, 1% w/v in DMEM) treated positive control (PC). For analysis, the NC (fresh medium) was set to 100%.
2.11. Multiphoton microscopy (MPM)
Imaging was performed using a custom built microscope system with a titanium–sapphire femtosecond laser (MaiTai XF1, Spectra Physics, Santa Clara, USA) as previously described.82 An excitation wavelength of 710 nm and a laser power of 18 mW was employed. The spectral emission filter ranged from 425 to 509 nm. For scaffold imaging, punches (∅ = 12 mm) of the scaffolds were put on Ibidi® (Ibidi GmbH, Planegg/Martinsried, Germany) glass bottom dishes (35 mm) and carefully pressed on the bottom with a cover glass before analysis. When imaging cross-linked scaffolds they were firstly rinsed with PBS 1×. HDFs morphology was assessed on glass bottom dishes (Ibidi®, 35 mm) with a density of 5 × 104 cells per dish. After 24 h, the medium was removed and 2 mL of fresh DMEM (+10% FCS) was added. When the cells were imaged on the scaffolds, HDFs were analyzed 24 hours after seeding. Before imaging, the electrospun scaffolds were removed from the inserts, flipped and carefully pressed on the bottom with a cover glass before analysis. To evaluate triple-helix content of GE in the electrospun scaffolds we defined a parameter (SAI, second harmonic to autofluorescence index, Scheme 3) similar to the second harmonic to autofluorescence aging index of dermis (SAAID).83 In this case, an excitation wavelength of 760 nm and a laser power of 18 mW was employed, while a 380/20 bandpass emission filter and a 435 nm long pass filter were used to detect second harmonic generation (SHG) and autofluorescence (AF), respectively. For the mean gray value intensity (GVI) analysis, ImageJ® was used as software to process and analyze the images.
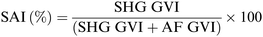 |
| | Scheme 3 Formula used to evaluate the second harmonic generation to autofluorescence index (SAI). | |
2.12. Fluorescence lifetime imaging microscopy (FLIM)
FLIM was performed to assess reduced (phosphorylated) nicotinamide adenine dinucleotide (NAD(P)H) or gelatin mean fluorescence lifetime using time correlated single photon counting (TCSPC) at an excitation wavelength of 710 nm and a laser power of 18 mW. Analyzed samples were handled as described for MPM in Section 2.11. FLIM data were recorded at an acquisition time of 180 s for 512 × 512 pixels with 64 time channels. The instrument response function was recorded using urea crystals (Sigma-Aldrich) at an excitation wavelength of 920 nm and a laser power of 4.5 mW for 120 s. The FLIM images were analyzed using the SPCImage software (Becker & Hickl GmbH, Berlin, Germany). A biexponential decay fitting model (Scheme 4) was employed at each pixel since NAD(P)H has two different lifetimes represented by τ1 and τ2.82,84 A χ2 < 1.1 was accepted for a good fitting. For gelatin analysis the mean fluorescence lifetime (τm, Scheme S1, ESI†) after the biexponential decay fitting was instead considered. A binning factor of 5 was used in the analysis. For each sample 4 different dishes were used and 6 images were acquired per dish.
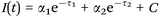 |
| | Scheme 4 Biexponential decay fitting used for the FLIM analysis. τ1 represents the free NAD(P)H lifetime time, while τ2 the protein bound NAD(P)H one. | |
2.13. Data analysis
All the reported graphs were plotted using Microsoft™ Excel. All data are presented as mean ± standard deviation (n = 4, unless stated otherwise in the materials and methods). Statistical significance was determined by a Student's two-tailed unpaired t-test. p ≤ 0.05 (*) was defined as statistically significant.
3. Results and discussion
3.1. Generation of electrospun scaffolds
For the production of the scaffolds, various electrospinning parameters were tested and those leading to a stable Taylor's cone and jet, as well as to smooth and uniform fibers, were then used throughout the study. The conditions were first defined for pure PLA and GE, and then adapted for the hybrid scaffolds. To date, some attempts have already been made to produce well-blended GE:PLA scaffolds.85–90 It has been demonstrated that HFP is a suitable solvent to process GE and PLA.85–89 By using HFP we were able to dissolve GE and PLA, either alone or together in different ratios, and to process the resulting blend solutions by electrospinning. Three different GE:PLA (w:w) ratios, 4:1, 5:2 and 1:1, were chosen for scaffolds production, characterization and comparison. Generally, we noticed that when using a concentration higher than 10% w/v for GE the electrospinning process was not stable and the solution jellified at room temperature. When using the two highest GE:PLA ratios (4:1 and 5:2), the relative GE concentration was 10% w/v, which is the threshold value that, according to our observations, avoided its jellification during the electrospinning experiment. For PLA we noticed that a final polymer concentration higher than 15% w/v led to instable electrospinning processes with inhomogeneous scaffolds. Cross-linking caused scaffold shrinkage, but this effect was more evident when having a higher GE content (Table 2). No significant shrinkage and weight loss occurred for pure PLA electrospun scaffolds (Table 2). Thus, the lower final GE content in the hybrid scaffolds is due to the cross-linking that leads to a water loss after intramolecular reaction (Fig. S1, ESI†) between GE aspartic (L-Asp)/glutamic (L-Glu) acidic residues and the free amines of GE lysines.52–54 Nevertheless, this GE loss is not significant (Table 2), so it was decided to always refer to the initial GE:PLA (w:w) ratio when characterizing the hybrid scaffolds.
Table 2 Composition of polymers and dimensions of the hybrid GE:PLA scaffolds, before and after cross-linking
| Initial GE:PLA w:w ratio |
Initial scaffold diameter (cm) |
GE:PLA final w:w ratio |
Final scaffold diameter (cm) |
| 4:1 |
9.0 |
18 (± 2):5 |
5.5 ± 0.5 |
| 5:2 |
9.0 |
9 (± 1):4 |
6.6 ± 0.4 |
| 1:1 |
9.0 |
9 (± 1):10 |
8.0 ± 0.2 |
| 0:1 |
9.0 |
0:1 |
9.0 ± 0.1 |
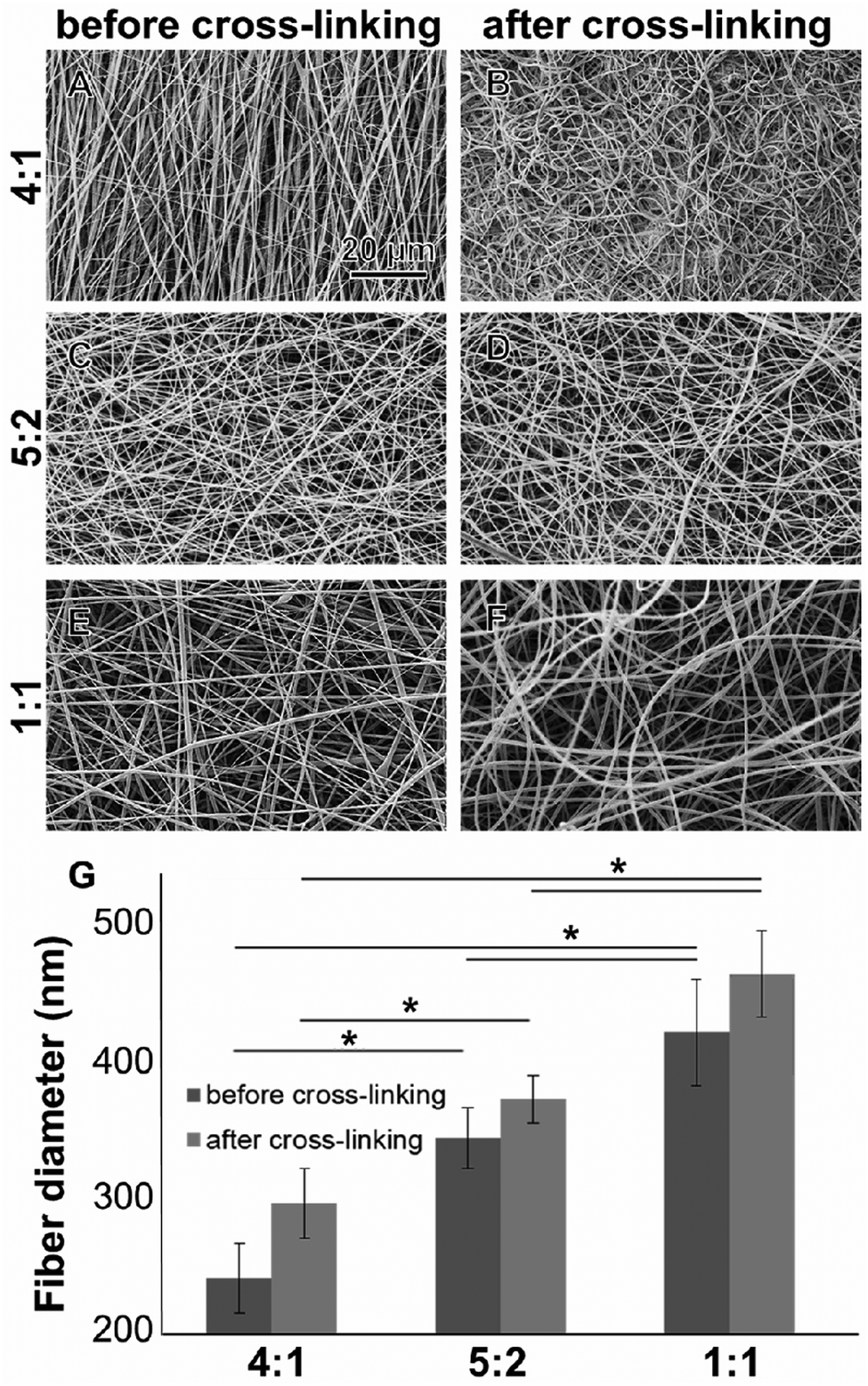
All electrospun scaffolds were characterized by SEM in order to assess the fiber quality after the electrospinning experiments and after the cross-linking reactions (Fig. 1 and Fig. S2, ESI†). Pure GE showed a homogeneous random-oriented fiber distribution (Fig. S2, ESI†), with a normally distributed fiber size around the mean value (Fig. S2B, ESI,† 334 ± 35 nm). After cross-linking, the highly porous structure, which is typical for electrospun scaffolds, was no longer visiable, and fiber size distributions could not be assessed (Fig. S2C, ESI†). Blend electrospun scaffolds often lack homogeneity,46,47 and the cross-linking reaction strongly alters their porous structure.54–56 When blending GE together with PLA in different ratios (4:1, 5:2 and 1:1) we were able to generate scaffolds with a random fiber orientation (Fig. 1A–F). In all conditions, we obtained uniform fibers with no beads showing a normal size distribution (Fig. S2D–I, ESI†). When increasing the PLA content, the mean fiber diameter of the hybrid scaffolds significantly increased (Fig. 1G) probably due to higher final polymer concentration and blend solution viscosity.85 The obtained values for the mean fiber diameters are reported in the ESI† together with the histograms (Fig. S2D–I, ESI†). After the cross-linking the mean fiber diameter generally increased but changes were not significant (in all cases p > 0.05). These results are in accordance with the data generated by Moon et al.,85 Wang et al.86 and Hoveizi et al.87 Kim et al.90 demonstrated that by using 2,2,2-trifluoroethanol as a solvent for the electrospinning experiments fiber sizes decrease with increasing PLA content. These results indicate that the choice of solvent plays an important role for polymers processing and needs to be carefully considered.85–90 Compared to pure GE, there are no significant morphological differences between the cross-linked and non-cross-linked PLA containing samples (Fig. 1). Non-cross-linked GE scaffolds dissolve in aqueous solutions. For tissue engineering applications however, non-water soluble scaffolds are mandatory. After cross-linking, all of the scaffolds were water resistant.
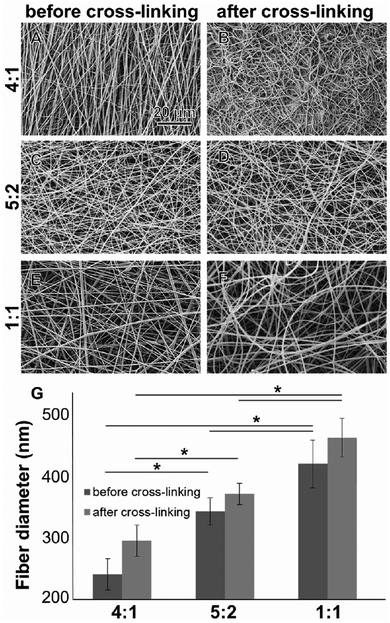 |
| | Fig. 1 (A–F) SEM images of electrospun GE:PLA at different ratios before (A, C and E) and after cross-linking (B, D and F). (G) Fiber diameter comparison among the hybrid GE:PLA electrospun scaffolds. *p ≤ 0.05. | |
3.2. Non-invasive assessment of cross-linking
In order to demonstrate cross-linking properties of the electrospun scaffold, we analyzed the generated GE:PLA scaffolds before and after cross-linking using Raman microspectroscopy. According to the obtained spectra, the region of interest was found to be between 740 and 920 cm−1. Particularly, the signals at 743 cm−1 and at 876 cm−1 (Fig. 4, downwards arrows) were attributed to vibrations of the free carboxylic groups of L-Glu91,92 and L-Asp,91,93 respectively. Both peaks were only present in the spectra of the uncross-linked GE containing scaffolds. After the cross-linking both signals disappeared, since L-Glu and L-Asp are involved in the formation of new amide bonds after the cross-linking reaction.45–47 These results indicate that the cross-linking process was successful. The signal at ca. 895 cm−1 (Fig. 4, asterisks) was instead assigned to the C–COO stretching94–96 of the polyester PLA, and so used to identify the increasing PLA content in the hybrid scaffolds. The relative intensity of this signal in the normalized spectra is higher the more PLA is present in the hybrid scaffolds (Fig. 2A). We performed PCA (Fig. 2B–G) to confirm these observations. Firstly, we were able to separately group the samples, before and after cross-linking (Fig. 2B). In this case, the two considered components for the loadings (Fig. 2C and D) both contained the peaks at 743 cm−1 and at 876 cm−1, which disappear after cross-linking. Thus, the cross-linked samples are grouped in the negative quarter of the correlation (Fig. 2B). The first considered component (Fig. 2C) includes the signal at 895 cm−1 reflecting the PLA content and so moving the hybrid samples towards the right with the increasing PLA percentage (Fig. 2B). Separately, PCA allowed us to detect the increasing PLA content in the final cross-linked hybrid scaffolds (Fig. 2E). In this case, pure PLA was used as reference, reflecting in the two considered components (Fig. 2F and G). The signal mainly contributing to the evaluation of the PLA content is the one at ca. 895 cm−1 as observed in the spectra (Fig. 2A), bringing pure PLA to the positive part of the correlation (Fig. 2E). The hybrid cross-linked samples are instead gradually shifted towards the negative quarter with the increasing GE percentage (Fig. 2E).
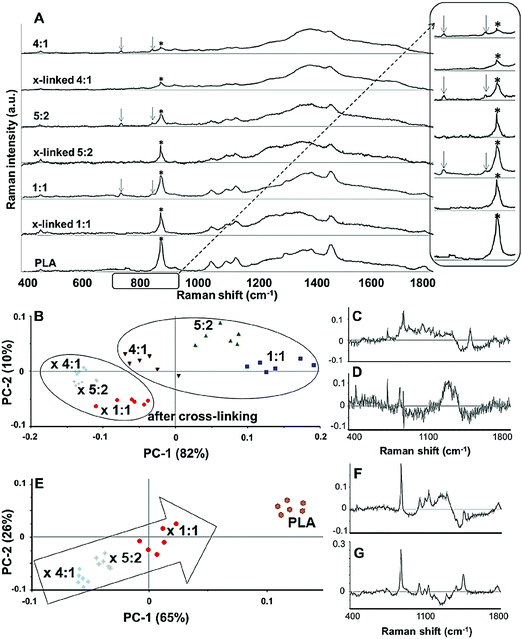 |
| | Fig. 2 (A) Raman spectra of GE:PLA hybrid electrospun scaffolds and of pure PLA, showing a peak for PLA (asterisks) and two peaks for cross-linking (downwards arrows). (B) Score graph after PCA analysis including all the hybrid GE:PLA scaffolds, showing a clear separation of cross-linked and uncross-linked samples. (C and D) Loadings for PC-1 (C) and for PC-2 (D); the wavenumbers with high (absolute) loadings are identified as belonging to uncross-linked GE (743 cm−1 and 876 cm−1) and PLA (895 cm−1). (E) Score graph after PCA analysis of the hybrid cross-linked GE:PLA scaffolds, showing the increase in PLA content (arrow). (F and G) Loadings for PC-1 (F) and for PC-2; the wavenumber with highest (absolute) loading is identified as belonging to PLA (895 cm−1). | |
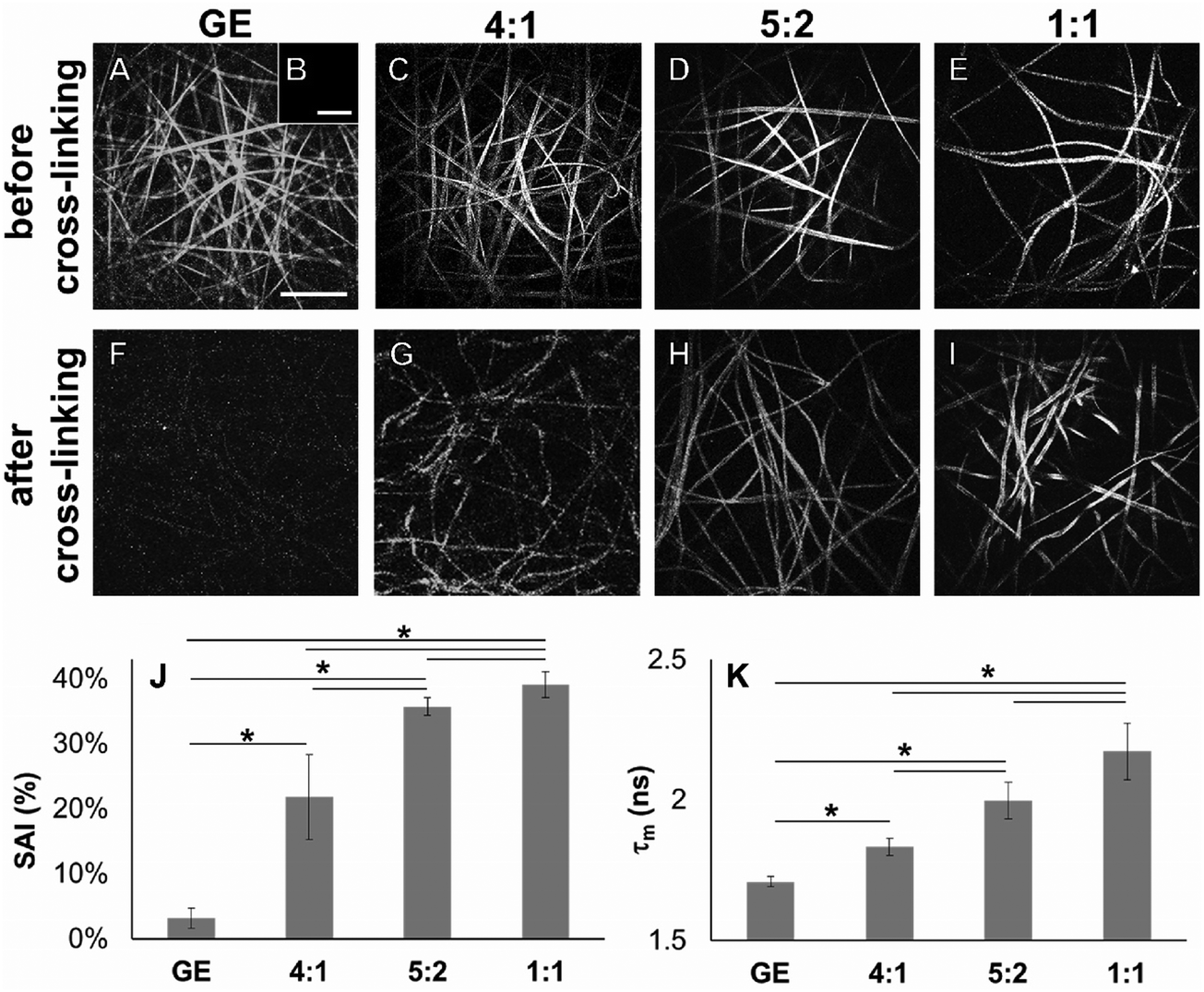
In addition to Raman microspectroscopy we employed MPM and FLIM to assess whether these methods can also be used to investigate proper cross-linking. Type B gelatin exhibits a broad fluorescence emission spectrum. By using two-photon excitation we were able to induce GE autofluorescence (AF) between 710 and 800 nm. As previously observed,97 pure electrospun PLA exhibits instead no AF (Fig. S3B, ESI†). Moreover, we were also able to detect a second harmonic generation (SHG) signal from the fibers in the GE scaffolds electrospun from HFP (Fig. 3F–J). Electrospun PLA fibers exhibit as expected no SHG (Fig. 3B). While the SHG signal for pure GE vanishes (Fig. 3F), the AF signal is stronger after cross-linking (Fig. S3F, ESI†). However, when imaging the hybrid scaffolds we noticed that the SHG signal in the cross-linked scaffolds could be more preserved when having a higher PLA content (Fig. 3G–I). According to these results we suggest that the presence of PLA helps to slow down the cross-linking reaction in the blend scaffolds helping to retain GE initial structure. To quantitatively evaluate this property we decided to compare the gray value intensities (GVI) of the SHG and AF images and defined the SHG to AF index (SAI). Before cross-linking, all hybrid scaffolds showed a SAI value, which was not significantly different from the one of pure electrospun GE (SAI GE: 41 ± 3%; Fig. S3J, ESI†). Pure cross-linked GE showed instead a SAI close to zero (SAI GE after cross-linking: 3 ± 1%; Fig. 3J). However, this parameter significantly increases for the hybrid cross-linked GE:PLA scaffolds with the increasing amount of PLA (SAI GE:PLA 4:1 = 22 ± 6% versus GE:PLA 5:2 = 36 ± 1%, p = 0.006; GE:PLA 5:2 = 36 ± 1% versus GE:PLA 1:1 = 39 ± 2%, p = 0.002; GE:PLA 1:1 = 39 ± 2% versus GE:PLA 4:1 = 22 ± 6%, p = 5 × 10−5; Fig. 3J). Interestingly, the hybrid cross-linked scaffold with an equal ratio between GE and PLA showed a SAI which was even not significantly different from the one of pure electrospun GE (GE, 41 ± 3% versus GE:PLA 1:1, 39 ± 2%, p = 0.37). It has been previously demonstrated by Bigi et al. that there is a direct relationship between the physical properties of GE structures and the triple-helix content, which decreases or even disappears after the cross-linking, depending on the used conditions.98 Similar results are reported for collagen after in vitro and in vivo measurements of the SHG-to-AF aging index of dermis (SAAID).83,99–101
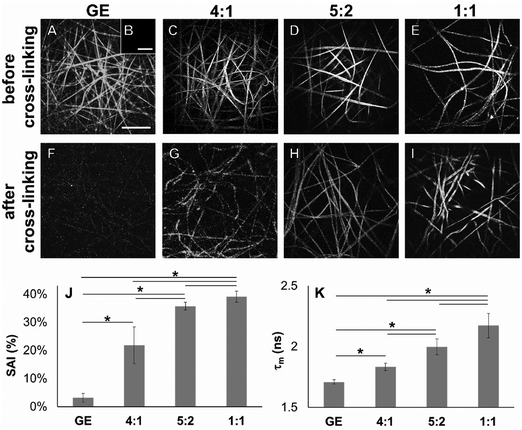 |
| | Fig. 3 (A–E) SHG images of non-cross-linked electrospun GE (A), PLA (B) and GE:PLA (C–E). (F–I) SHG images of cross-linked electrospun GE (F) and GE:PLA (G–I). (J) SHG to AF index (SAI) of GE-containing electrospun cross-linked scaffolds after two-photon excitation at 760 nm. (K) Mean fluorescence lifetime (τm) of GE-containing electrospun cross-linked scaffolds after two-photon excitation at 710 nm. Scale bars equal 30 μm. *p ≤ 0.05. | |
Using FLIM, we evaluated the fluorescence lifetimes of the cross-linked hybrid scaffolds and compared them to those of pure GE scaffolds, either before (Fig. S3K, ESI†) or after the cross-linking process (Fig. 3K). To compare the decay time of the scaffolds, the mean fluorescence lifetime (τm, Scheme S1, ESI†) after a biexponential decay curve fitting was considered. Before cross-linking, all hybrid scaffolds showed a τm which is not significantly different from the one of electrospun GE (τm GE = 2.33 ± 0.06 ns; Fig. S3K, ESI†). τm significantly decreases for GE after the cross-linking (GE = 1.71 ± 0.02 ns, p = 3 × 10−6). When analyzing the cross-linked hybrid scaffolds we could observe that τm significantly increases for the hybrid scaffolds with the increasing PLA percentage. Specifically, the higher the PLA percentage, the closer the τm is to the one of uncross-linked GE (GE:PLA 1:1 = 2.17 ± 0.11 ns versus 5:2 = 1.99 ± 0.07 ns, p = 0.004; 1:1 = 2.17 ± 0.11 ns versus 4:1 = 1.83 ± 0.03 ns, p = 7 × 10−4; 5:2 = 1.99 ± 0.07 ns versus 4:1 = 1.83 ± 0.03 ns, p = 0.012; Fig. 3K). Moreover, in all cases, τm was at the same time significantly longer when compared to the cross-linked GE. These results show that by blending GE with PLA in electrospun scaffolds it is possible to partially retain the initial GE τm even in the cross-linked samples (1:1 = 2.17 ± 0.11 ns versus GE = 1.71 ± 0.02 ns, p = 0.004; 5:2 = 1.99 ± 0.07 ns versus GE = 1.71 ± 0.02 ns, p = 0.014; 4:1 = 1.83 ± 0.03 ns versus GE = 1.71 ± 0.02 ns, p = 0.026). However, for all the hybrid cross-linked scaffolds, the mean fluorescence lifetime τm was still significantly shorter when compared to the uncross-linked GE (1:1 = 2.17 ± 0.11 ns versus GE = 2.33 ± 0.06 ns, p = 0.037; 5:2 = 1.99 ± 0.07 ns versus GE = 2.33 ± 0.06 ns, p = 0.002; 4:1 = 1.83 ± 0.03 ns versus GE = 2.33 ± 0.06 ns, p = 6 × 10−5). To summarize, we were able to evaluate structural properties of GE-containing electrospun scaffolds in a fast and non-destructive way using MPM and FLIM techniques. Nevertheless, differential scanning calorimetry and X-ray diffraction may be suitable analyses to evaluate the triple-helix content in the electrospun scaffolds and correlate the results to our observations.98
3.3. Impact of various GE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :PLA ratios on the mechanical properties
:PLA ratios on the mechanical properties
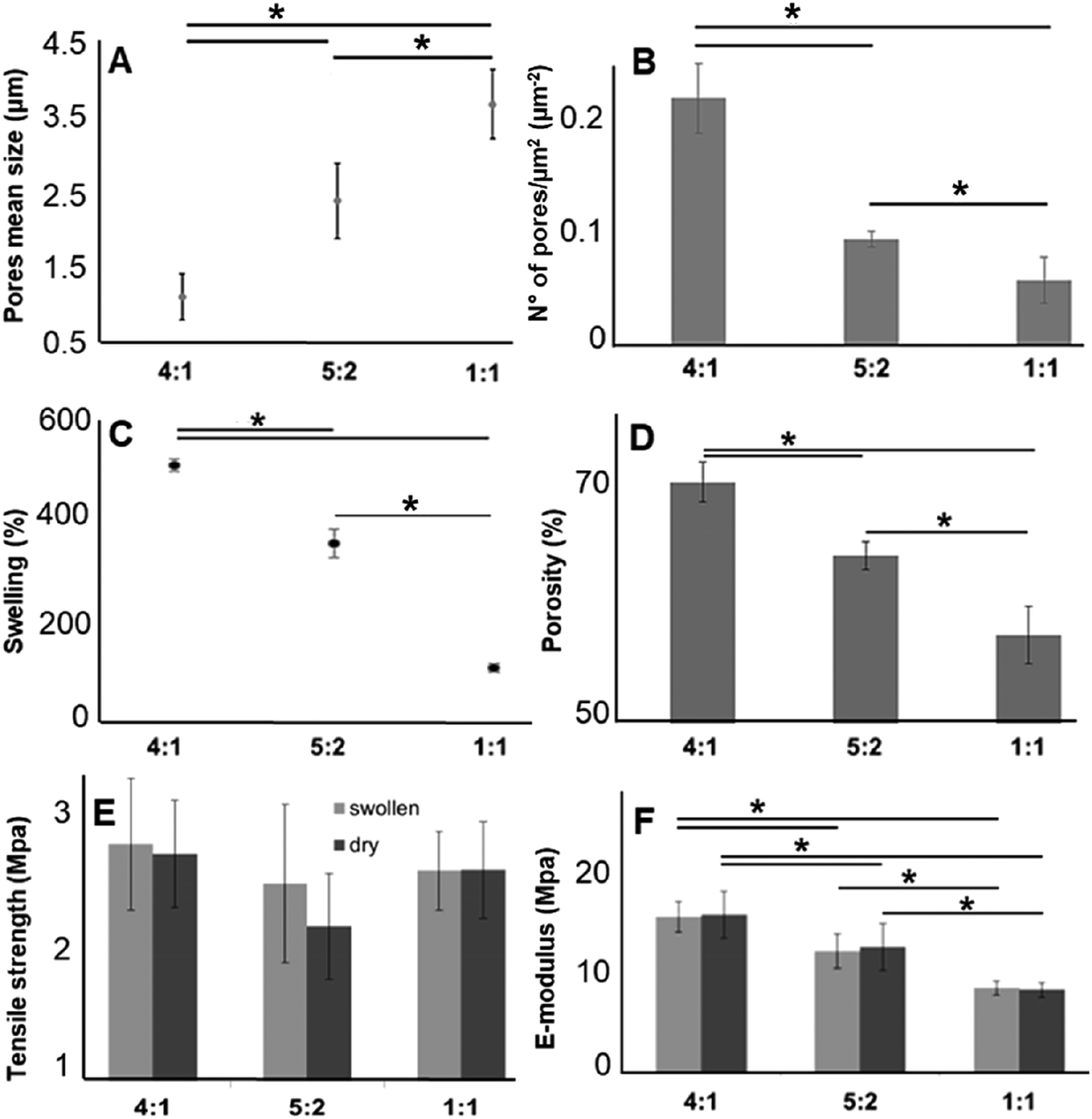
As described above, for tissue engineering applications water insoluble scaffolds are mandatory. After we demonstrated proper cross-linking, we used the cross-linked and non-soluble samples for further investigations. According to the scaffold morphology (Fig. 1) the cross-linked hybrid scaffolds with a higher GE content showed a higher porosity. This observation could be confirmed with DiameterJ analysis of the SEM images. According to this analysis, the mean pore size increased with an increasing amount of PLA (Fig. 4A), while the number of pores (expressed as number of pores per μm2; Fig. 4B) decreased with increasing PLA content. These microscopical properties of the hybrid cross-linked scaffolds correlate with the results obtained with the swelling test and the liquid displacement method to determine their wettability (Fig. 4C) and porosity (Fig. 4D), respectively. According to our observations, the scaffolds with higher GE content and a greater number of pores retained more water (Fig. 4C). These results are in accordance with previous studies.99–101 Furthermore, the amount of GE is correlated with the resulting scaffold porosity (Fig. 4D). Moreover, we could also infer that a higher porosity reflects in a higher amount of water retained by the cross-linked hybrid scaffolds. As previously demonstrated, these swelling properties are also directly related to the GE content.102,103 The scaffolds wettability was also evaluated by measuring the contact angle. Both 4:1 and 5:2 GE:PLA scaffolds were highly hydrophilic, while the 1:1 GE:PLA blend was highly hydrophobic. The obtained contact angle values were of 0° for the first two scaffolds (4:1 and 5:2) and 129° ± 3° for the 1:1 GE:PLA blend. This value is very close to the one of pure PLA.97
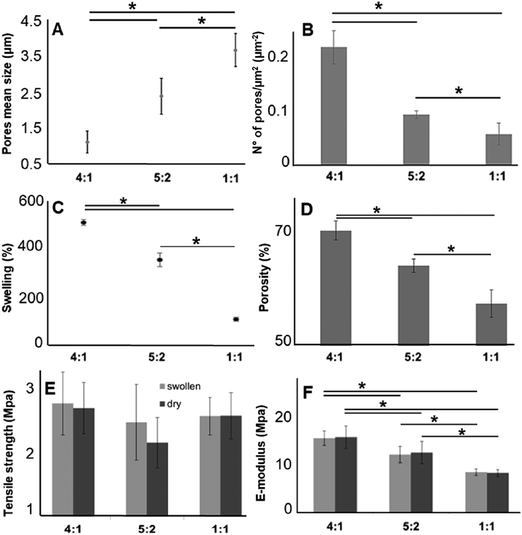 |
| | Fig. 4 Comparison of the mechanical properties of the cross-linked hybrid GE:PLA scaffolds. (A) Pores mean size and (B) mean number of pores divided per the scaffold surface. Both calculated from SEM images. (C) Swelling properties. (D) Porosity percentage according to the liquid displacement method. (E and F) Comparison of tensile strength (E) and E-modulus (F) among the cross-linked scaffolds with different GE:PLA ratios before and after swelling. *p ≤ 0.05. | |
Furthermore, we investigated the tensile strength and the elastic modulus (E-modulus), before and after swelling, for cross-linked hybrid GE:PLA scaffolds (Fig. 4E and F) as well as for pure electrospun PLA and cross-linked GE. In accordance with previous studies104–106 all hybrid scaffolds show a much greater elongation at break after swelling (elongation (%) > 100%; Fig. S4, ESI†). However, no significant differences could be determined when comparing the same hybrid scaffolds, before and after swelling in both, tensile strength (Fig. 4E) and E-modulus (Fig. 4F). Jing et al. described the same characteristics when blending GE with polypropylene carbonate.106 We observed the same behavior for the E-modulus of pure PLA (PLA before swelling: 26.8 ± 4.5 MPa versus PLA after swelling: 27.1 ± 3.2 MPa; p = 0.62). Pure cross-linked GE was instead significantly more elastic after swelling (E-modulus GE before swelling: 26.6 ± 2.2 MPa versus GE after swelling: 1.22 ± 0.34 MPa, p = 3 × 10−6). According to our measurements, the tensile strength of the hybrid scaffolds is not significantly affected by the different polymer ratios (Fig. 4E). Generally, the obtained values for the tensile strength of the hybrid scaffolds are close to the one of pure PLA (PLA before swelling: 1.88 ± 0.2 MPa; PLA after swelling: 1.94 ± 0.21 MPa) and cross-linked GE (GE before swelling: 2.63 ± 0.54 MPa; GE after swelling: 2.12 ± 0.31 MPa). In contrast, the E-modulus depends on the PLA content. The higher the PLA amount in the scaffolds, the higher the E-modulus in both conditions, dry and after swelling (Fig. 4F). In general, GE stiffness increases after its cross-linking.52–56 As already observed with MPM and FLIM analyses, we believe that the presence of PLA helps to slow down the cross-linking reaction. Thus, the higher the PLA percentage the more elastic result the final hybrid cross-linked scaffolds. With these results, we clearly demonstrated that wettability, porosity but also the stiffness of scaffold can be tuned by varying the ratio of synthetic and natural polymers. However, E-modulus of pure cross-linked electrospun GE may represent a threshold value when aiming to increase/decrease scaffold stiffness, as it showed the lowest elasticity when measured dry, as well as the highest one after swelling.
3.4. Cell–material interactions and biocompatibility
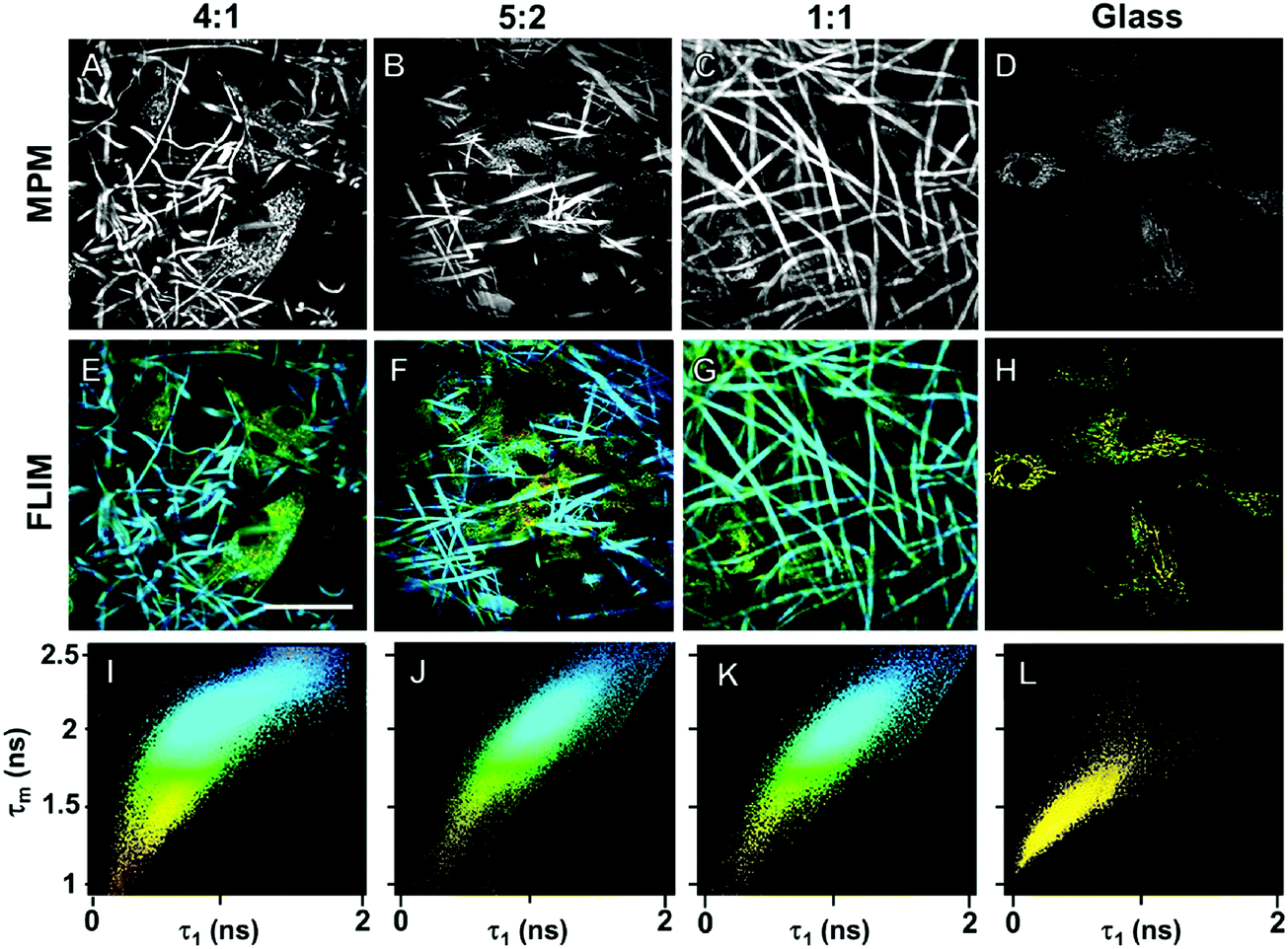
Biocompatibility of the GE-containing scaffolds was confirmed employing an MTS proliferation assay (Fig. S5, ESI†). According to the ISO 10993-5 standard, a proliferation higher than 80% indicates no cytotoxic effects. According to the results, neither the cross-linking reaction nor the polymers have cytotoxic effects on HDFs. We demonstrated the biocompatibility of pure PLA in a previous work.97 In order to detect cellular adhesion and growth of human dermal fibroblasts (HDFs) on the different hybrid electrospun scaffolds, we employed MPM, which represents a non-invasive, marker-free methodology.107–110 All the cross-linked hybrid GE:PLA scaffolds with a different ratio between the two polymers were investigated. MPM images of electrospun scaffolds seeded with HDFs are presented in Fig. 5. Gelatin is reported to exhibit autofluorescence at an excitation wavelength of 355 nm.111 Thus, using a two photon excitation, we detected an autofluorescence signal at a wavelength of 710 nm. By 710 nm we were also able to image HDFs within the scaffolds, since NAD(P)H autofluorescence occurs at the same wavelength (Fig. 5A–D).83 Interestingly, these images clearly show that HDFs adhere not only on the scaffold surface, but mainly within the fibrous structure.
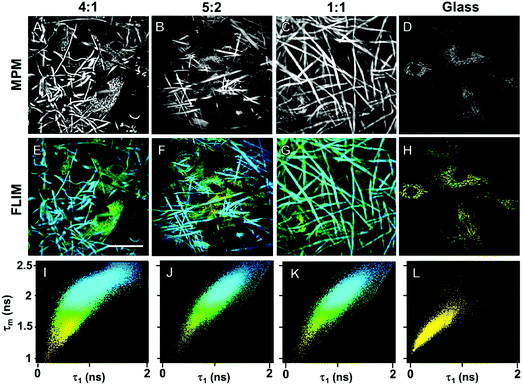 |
| | Fig. 5 (A–D) MPM imaging of HDFs seeded on hybrid cross-linked GE:PLA scaffolds (A–C) or on glass (D). (E–H) FLIM false color coded imaging of HDFs seeded on hybrid GE:PLA scaffolds (E–G) or on glass (H) after biexponential decay fitting. (I–L) 2D-correlation between τ1 and τm after biexponential decay curve fitting of the FLIM data. Scale bar equals 25 μm. | |
Furthermore, after MPM imaging of HDFs within the fibrous hybrid GE:PLA scaffolds, we investigated the same samples using FLIM (Fig. 5E–J), which allows the analysis of the metabolism of the target cells.112 Interestingly, despite the strong autofluorescence of GE, we were able to distinguish cells from the scaffolds by a false color-coding, based on a 2D-correlation using a biexponential decay curve fitting. The cells show a much shorter τ1 than the scaffolds, due to the fluorescence lifetime of free NAD(P)H.83,112 This value also leads to a shorter mean fluorescence lifetime (τm), which is directly correlated to the τ1 values.113 Shorter τ1 and τm values were colored yellow, and longer τ1s and τms were colored blue (Fig. 5E–G). Looking at the 2D-correlation, where τ1 and τm are plotted as variables on the x and y axes (Fig. 5I–K), the pixels obtained from the cells lay in the lower left part of the plot, while those due to scaffold contribution in the upper right one. When analyzing HDFs on glass (Fig. 5H), all the values lay in the lower left part of the plot, with more than the 95% of the points having τ1 < 1000 ps and τm < 1750 ps (Fig. 5L). To conclude, we successfully demonstrated that the non-invasive methods MPM and FLIM could be used to display cells in and on electrospun scaffolds. Cells interact with and migrate into the electrospun scaffolds.
4. Conclusions
In this work, we demonstrated how different GE:PLA ratios impact the characteristics and mechanical properties of electrospun scaffolds. GE and PLA are both bioresorbable and their properties vary depending on their molecular weight and the manufacturing technique. Thus, after properly choosing the starting materials and their processing conditions, it is possible to obtain electrospun blend scaffolds with unique features. By changing the ratio between the natural (here GE) and the synthetic (here PLA) polymer, we were able to obtain scaffolds with different porosities, which directly correlate with their swelling properties. In particular, the porosity and the swelling properties of the scaffolds increased when using a higher GE content, so were their stiffness profiles. Furthermore, we implemented non-invasive methods to characterize electrospun scaffolds. Raman microspectroscopy was used to assess proper cross-linking of the scaffold, while we employed MPM and FLIM to non-invasively visualize and monitor cells on and in the electrospun scaffolds. We were able to discriminate the signals of cellular NAD(P)H of HDFs from the signals of the scaffolds, which will enable the online in vitro detection of the glycolytic activity in future applications. To conclude, we were able to tune the properties of hybrid GE:PLA electrospun scaffolds, and we characterized these scaffolds in a non-destructive, marker-independent way.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank Simone Liebscher (University Women's Hospital Tübingen, Germany) and Alessandro Laurita (University of Basilicata, Italy) for their technical support. Special thanks are due to Shannon L. Layland (University Women's Hospital Tübingen, Germany) for his helpful comments. The authors are grateful for the financial support by the DAAD (Year scholarship program 57130104 to GP), as well as the Deutsche Forschungsgemeinschaft (SCHE701/14-1, INST 2388/30-1) and the Ministry of Science, Research and Arts of Baden Württemberg (Az.: SI-BW 01222-91, 33-729.55-3/214) (all to KS-L).
References
- C. Frantz, K. M. Stewart and V. M. Weaver, The extracellular matrix at a glance, J. Cell Sci., 2010, 123, 4195–4200, DOI:10.1242/jcs.023820.
- B. Yue, Biology of the Extracellular Matrix: An Overview, J. Glaucoma, 2014, S20–S23, DOI:10.1097/IJG.0000000000000108.
- M. Martins-Green and M. J. Bissell, Cell-ECM interactions in development, Semin. Dev. Biol., 1995, 6, 149–159, DOI:10.1016/S1044-5781(06)80024-1.
- F. Rosso, A. Giordano, M. Barbarisi and A. Barbarisi, From Cell–ECM interactions to tissue engineering, J. Cell. Physiol., 2004, 199, 174–180, DOI:10.1002/jcp.10471.
- B. R. Freedman, N. D. Bade, C. N. Riggin, S. Zhang, P. G. Haines, K. L. Ong and P. A. Janmey, The (dys)functional extracellular matrix, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 3153–3164, DOI:10.1016/j.bbamcr.2015.04.015.
- A. P. Liu, O. Chaudhuri and S. H. Parekh, New advances in probing cell–extracellular matrix interactions, Integr. Biol., 2017, 9, 383–405, 10.1039/C6IB00251J.
- S. Hinderer, S. Lee Layland and K. Schenke-Layland, ECM and ECM-like materials-biomaterials for applications in regenerative medicine and cancer therapy, Adv. Drug Delivery Rev., 2016, 97, 260–269, DOI:10.1016/j.addr.2015.11.019.
- J. Gunn and M. Zhang, Polyblend nanofibers for biomedical applications: perspectives and challenges, Trends Biotechnol., 2010, 28, 189–197, DOI:10.1016/j.tibtech.2009.12.006.
- N. Bhattarai, Z. Li, J. Gunn, M. Leung, A. Cooper, D. Edmondson, O. Veiseh, M. H. Chen, Y. Zhang, R. G. Ellenbogen and M. Zhang, Natural-Synthetic Polyblend Nanofibers for Biomedical Applications, Adv. Mater., 2009, 21, 2792–2797, DOI:10.1002/adma.200802513.
- A. Espíndola-González, A. L. Martínez-Hernández and F. Fernández-Escobar,
et al., Natural-Synthetic Hybrid Polymers Developed via Electrospinning: The Effect of PET in Chitosan/Starch System, Int. J. Mol. Sci., 2011, 12, 1908–1920, DOI:10.3390/ijms12031908.
- L. Martinova and D. Lubasova, Reasons for using polymer blends in the electrospinning process, AIP Conf. Proc., 2012, 1502, 115–128, DOI:10.1063/1.4769138.
- P. Tipduangta, P. Belton, L. Fábián, L. Ying Wang, H. Tang, M. Eddleston and S. Qi, Electrospun Polymer Blend Nanofibers for Tunable Drug Delivery: The Role of Transformative Phase Separation on Controlling the Release Rate, Mol. Pharmaceutics, 2016, 13, 25–39, DOI:10.1021/acs.molpharmaceut.5b00359.
- S. Heydarkhan-Hagvall, K. Schenke-Layland, A. P. Dhanasopon, F. Rofail, H. Smith, B. M. Wu, R. Shemin, R. E. Beygui and W. R. MacLellan, Three-dimensional electrospun ECM-based hybrid scaffolds for cardiovascular tissue engineering, Biomaterials, 2008, 29, 2907–2914, DOI:10.1016/j.biomaterials.2008.03.034.
- X. Wang, B. Ding and B. Li, Biomimetic electrospun nanofibrous structures for tissue engineering, Mater. Today, 2013, 16, 229–241, DOI:10.1016/j.mattod.2013.06.005.
- W. Liu, S. Thomopoulos and Y. Xia, Electrospun Nanofibers for Regenerative Medicine, Adv. Healthcare Mater., 2012, 1, 10–25, DOI:10.1002/adhm.201100021.
- S. Lyu, C. Huang, H. Yang and X. Zhang, Electrospun Fibers as a Scaffolding Platform for Bone Tissue Repair, J. Orthop. Res., 2013, 31, 1382–1389, DOI:10.1002/jor.22367.
- P. P. Bonvallet, M. J. Schultz and E. H. Mitchell,
et al., Microporous Dermal-Mimetic Electrospun Scaffolds Pre-Seeded with Fibroblasts Promote Tissue Regeneration in Full-Thickness Skin Wounds, PLoS One, 2015, 10, e0122359, DOI:10.1371/journal.pone.0122359.
- R. Khajavi and M. Abbasipour, Electrospinning as a versatile method for fabricating coreshell, hollow and porous nanofibers, Sci. Iran., 2012, 19, 2029–2034, DOI:10.1016/j.scient.2012.10.037.
- A. P. Kishan and E. M. Cosgriff-Hernandez, Recent advancements in electrospinning design for tissue engineering applications: a review, J. Biomed. Mater. Res., Part A, 2017, 105, 2892–2905, DOI:10.1002/jbm.a.36124.
- G. H. Lee, J. C. Song and K. B. Yoon, Controlled wall thickness and porosity of polymeric hollow nanofibers by coaxial electrospinning, Macromol. Res., 2010, 18, 571–576, DOI:10.1007/s13233-010-0607-9.
- Q. L. Loh and C. Choong, Three-Dimensional Scaffolds for Tissue Engineering Applications: Role of Porosity and Pore Size, Tissue Eng., Part B, 2013, 19, 485–502, DOI:10.1089/ten.teb.2012.0437.
- H. G. Sundararaghavan, R. B. Metter and J. A. Burdick, Electrospun Fibrous Scaffolds with Multi-scale and Photopatterned Porosity, Macromol. Biosci., 2010, 10, 265–270, DOI:10.1002/mabi.200900363.
- J. Rnjak-Kovacina and A. S. Weiss, Increasing the pore size of electrospun scaffolds, Tissue Eng., Part B, 2011, 17, 365–372, DOI:10.1089/ten.teb.2011.0235.
- B. A. Blakeney, A. Tambralli and J. M. Anderson,
et al., Cell Infiltration and Growth in a Low Density, Uncompressed Three-Dimensional Electrospun Nanofibrous Scaffold, Biomaterials, 2011, 32, 1583–1590, DOI:10.1016/j.biomaterials.2010.10.056.
- K. Kim, M. Yu, X. Zong, J. Chiu, D. Fang, Y. Seo, B. S. Hsiao, B. Chu and M. Hadjiargyrou, Control of degradation rate and hydrophilicity in electrospun non-woven poly(D,L-lactide) nanofiber scaffolds for biomedical applications, Biomaterials, 2003, 24, 4977–4985, DOI:10.1016/S0142-9612(03)00407-1.
- S. Ramakrishna, K. Fujihara, W. Teo, T. Yong, Z. Ma and R. Ramaseshan, Electrospun nanofibers: solving global issues, Mater. Today, 2006, 9, 40–50, DOI:10.1016/S1369-7021(06)71389-X.
- Q. P. Pham, U. Sharma and A. G. Mikos, Electrospinning of Polymeric Nanofibers for Tissue Engineering Applications: A Review, Tissue Eng., 2006, 12, 1197–1211, DOI:10.1089/ten.2006.12.1197.
- A. Greiner and J. H. Wendorff, Electrospinning: A Fascinating Method for the Preparation of Ultrathin Fibers, Angew. Chem., Int. Ed., 2007, 46, 5670–5703, DOI:10.1002/anie.200604646.
-
L. Xiao, B. Wang, G. Yang and M. Gauthier, Poly(Lactic Acid)-Based Biomaterials: Synthesis, Modification and Applications, in Biomedical Science, Engineering and Technology, ed. D. N. Ghista, 2012, pp. 247–282 Search PubMed.
- Y. Cheng, S. Deng, P. Chen and R. Ruan, Polylactic acid (PLA) synthesis and modification: a review, Front. Chem. China, 2009, 4, 259–264 CrossRef.
- B. Gupta, N. Revagade and J. Hilborn, Poly(lactic acid) fiber: an overview, Prog. Polym. Sci., 2007, 32, 455–482 CrossRef.
- E. D. Boland, G. E. Wnek, D. G. Simpson, K. J. Pawlowski and G. L. Bowlin, Tailoring tissue engineering scaffolds using electrostatic processing techniques: a study of poly(glycolic acid) electrospinning, J. Macromol. Sci., Part A: Pure Appl. Chem., 2001, 38, 1231–1243 CrossRef.
- P. Gentile, V. Chiono, I. Carmagnola and P. V. Hatton, An Overview of Poly(lactic-co-glycolic) Acid (PLGA)-Based Biomaterials for Bone Tissue Engineering, Int. J. Mol. Sci., 2014, 15, 3640–3659, DOI:10.3390/ijms15033640.
- Y. Dong, S. Liao, M. Ngiam, C. K. Chan and S. Ramakrishna, Degradation Behaviors of Electrospun Resorbable Polyester Nanofibers, Tissue Eng., Part B, 2009, 15, 333–351, DOI:10.1089/ten.teb.2008.0619.
- H. Liu, S. Wang and N. Qi, Controllable structure, properties, and degradation of the electrospun PLGA/PLA-blended nanofibrous scaffolds, J. Appl. Polym. Sci., 2012, 125, E468–E476, DOI:10.1002/app.36757.
- W. J. Li,
et al., Electrospun nanofibrous structure: a novel scaffold for tissue engineering, J. Biomed. Mater. Res., 2002, 60, 613–621 CrossRef PubMed.
- S. Khorshidi, A. Solouk, H. Mirzadeh, S. Mazinani, J. M. Lagaron, S. Sharifi and S. Ramakrishna, A review of key challenges of electrospun scaffolds for tissue-engineering applications, J. Tissue Eng. Regener. Med., 2016, 10, 715–738, DOI:10.1002/term.1978.
- J. A. Matthews,
et al., Electrospinning of collagen nanofibers, Biomacromolecules, 2002, 3, 232–238 CrossRef PubMed.
- M. Li,
et al., Electrospun protein fibers as matrices for tissue engineering, Biomaterials, 2005, 26, 5999–6008 CrossRef PubMed.
- J. D. Schiffman and C. Schauer, A Review: Electrospinning of Biopolymer Nanofibers and their Applications, Polym. Rev., 2008, 48, 317–352, DOI:10.1080/15583720802022182.
- J. M. Dang and K. W. Leong, Natural polymers for gene delivery and tissue engineering, Adv. Drug Delivery Rev., 2006, 58, 487–499 CrossRef PubMed.
- D. I. Zeugolis and S. T. Khew,
et al., Electro-spinning of pure collagen nano-fibres – just an expensive way to make gelatin?, Biomaterials, 2008, 29, 2293–2305, DOI:10.1016/j.biomaterials.2008.02.009.
- O. Evrova,
et al., Hybrid Randomly Electrospun Poly(lactic-co-glycolic acid):Poly(ethylene oxide) (PLGA:PEO) Fibrous Scaffolds Enhancing Myoblast Differentiation and Alignment, ACS Appl. Mater. Interfaces, 2016, 8, 31574–31586, DOI:10.1021/acsami.6b11291.
- Y. Liu,
et al., Engineering of bio-hybrid materials by electrospinning polymer-microbe fibers, PNAS, 2009, 106, 14201–14206, DOI:10.1073/pnas.0903238106.
- A. Sionkowska, Current research on the blends of natural and synthetic polymers as new biomaterials: review, Prog. Polym. Sci., 2011, 36, 1254–1276, DOI:10.1016/j.progpolymsci.2011.05.003.
- K. Kwon and T. Matsuda, Co-Electrospun Nanofiber Fabrics of Poly(l-lactide-co-ε-caprolactone) with Type I Collagen or Heparin, Biomacromolecules, 2005, 6, 2096–2105, DOI:10.1021/bm050086u.
- Y. Zhang, B. Su, J. Venugopal, S. Ramakrishna and C. Lim, Biomimetic and bioactive nanofibrous scaffolds from electrospun composite nanofibers, Int. J. Nanomed., 2007, 2, 623–638 Search PubMed.
- J. E. Eastoe, The amino acid composition of mammalian collagen and gelatin, Biochem. J., 1955, 61, 589–600 CrossRef PubMed.
-
R. T. Jones, Gelatin: Manufacture and Physio-Chemical Properties, in Pharmaceutical Capsules, ed. F. Podczeck and B. E. Jones, Pharmaceutical Press, London, UK, 2004, pp. 23–60 Search PubMed.
- H. B. Bohidar and S. S. Jena, Study of sol-state properties of aqueous gelatin solutions, J. Chem. Phys., 1994, 100, 6888–6895 CrossRef.
- A. L. Butcher, C. T. Koh and M. L. Oyen, Systematic mechanical evaluation of electrospun gelatin meshes, J. Mech. Behav. Biomed. Mater., 2017, 69, 412–419, DOI:10.1016/j.jmbbm.2017.02.007.
- J. B. Rose, S. Pacelli, A. J. E. Haj, H. S. Dua, A. Hopkinson, L. J. White and F. Rose, Gelatin-Based Materials in Ocular Tissue Engineering, Materials, 2014, 7, 3106–3135, DOI:10.3390/ma7043106.
- K. Tomihata and Y. Ikada, Cross-Linking of Gelatin with Carbodiimides, Tissue Eng., 2007, 2, 307–313, DOI:10.1089/ten.1996.2.307.
- K. Sisson,
et al., Evaluation of Cross-Linking Methods for Electrospun Gelatin on Cell Growth and Viability, Biomacromolecules, 2009, 10, 1675–1680, DOI:10.1021/bm900036s.
- G. A. Digenis,
et al., Cross-Linking of Gelatin Capsules and Its Relevance to Their in Vitro-in Vivo Performance, J. Pharm. Sci., 1994, 83, 915–921, DOI:10.1002/jps.2600830702.
- C. R. Cammarata, M. E. Hughes and C. M. Ofner, Carbodiimide Induced Cross-Linking, Ligand Addition, and Degradation in Gelatin, Mol. Pharmaceutics, 2015, 12, 783–793, DOI:10.1021/mp5006118.
- M. V. Natu, J. P. Sardinha, I. J. Correia and M. H. Gil, Controlled release gelatin hydrogels and lyophilisates with potential application as ocular
inserts, Biomed. Mater., 2007, 2, 241–249 CrossRef PubMed.
- J. Y. Lai and A. C. Hsieh, A gelatin-g-poly(N-isopropylacrylamide) biodegradable in situ gelling delivery system for the intracameral administration of pilocarpine, Biomaterials, 2012, 33, 2372–2387 CrossRef PubMed.
- D. Jain, E. Carvalho, A. K. Banthia and R. Banerjee, Development of polyvinyl alcohol-gelatin membranes for antibiotic delivery in the eye, Drug Dev. Ind. Pharm., 2011, 37, 167–177 CrossRef PubMed.
- W. M. Hsu, K. H. Chen, J. Y. Lai and G. H. Hsiue, Transplantation of human corneal endothelial cells using functional biomaterials: poly(N-isopropylacrylamide) and gelatin, J. Exp. Clin. Med., 2013, 5, 56–64 CrossRef.
- Y. Katagiri, S. A. Brew and K. C. Ingham, All six modules of the gelatin-binding domain of fibronectin are required for full affinity, J. Biol. Chem., 2003, 278, 11897–11902 CrossRef PubMed.
- J. Y. Lai, Y. T. Li, C. H. Cho and T. C. Yu, Nanoscale modification of porous gelatin scaffolds with chondroitin sulfate for corneal stromal tissue engineering, Int. J. Nanomed., 2012, 7, 1101–1114 CrossRef PubMed.
- C. Tondera, S. Hauser and A. Krüger-Genge,
et al., Gelatin-based Hydrogel Degradation and Tissue Interaction in vivo: Insights from Multimodal Preclinical Imaging in Immunocompetent Nude Mice, Theranostics, 2016, 6, 2114–2128, DOI:10.7150/thno.16614.
- K. Park, Y. M. Ju, J. S. Son, K. Ahn and D. K. Han, Surface modification of biodegradable electrospun nanofiber scaffolds and their interaction with fibroblasts, J. Biomater. Sci., Polym. Ed., 2007, 18, 369–382, DOI:10.1163/156856207780424997.
- M. Aoki, S. Miyamoto, K. Okamura, T. Yamashita, Y. Ikada and S. Matsuda, Tensile properties and biological response of poly(L-lactic acid) felt graft: an experimental trial for rotator-cuff reconstruction, J. Biomed. Mater. Res., Part B, 2004, 71, 252–259, DOI:10.1002/jbm.b.30084.
-
W. Groot, J. Van Krieken, O. Sliekersl and S. De Vos, Production and Purification of Lactic Acid and Lactide, in Poly(Lactic Acid): Synthesis, Structures, Properties, Processing, and Applications, ed. R. Auras, L. Lim, S. Selke and H. Tsuji, Wiley-VCH, Weinheim, Germany, 2010, pp. 1–18, DOI:10.1002/9780470649848.
-
J. M. Raquez, R. Mincheva, O. Coulembier and P. Dubois, Polymer Science: A Comprehensive Reference, 2012, vol. 1, pp. 761–778 Search PubMed.
-
A. Södergård and M. Stolt, Industrial Production of High Molecular Weight Poly(Lactic Acid), in Poly(Lactic Acid): Synthesis, Structures, Properties, Processing, and Applications, ed. R. Auras, L. Lim, S. Selke and H. Tsuji, Wiley-VCH, Weinheim, Germany, 2010, pp. 27–41, DOI:10.1002/9780470649848.
- D. Garlotta, A Literature Review of Poly(Lactic Acid), J. Polym. Environ., 2001, 9, 63–84, DOI:10.1023/A:1020200822435.
- A. Södergård and M. Stolt, Properties of lactic acid based polymers and their correlation with composition, Prog. Polym. Sci., 2002, 27, 1123–1163, DOI:10.1016/s0079-6700(02)00012-6.
- S. H. Lee and W. S. Song, Enzymatic Hydrolysis of Polylactic Acid Fiber, Appl. Biochem. Biotechnol., 2011, 164, 89–102, DOI:10.1007/s12010-010-9117-7.
- J. E. Bergsma, F. R. Rozema and R. M. Bos,
et al., Biocompatibility and degradation mechanisms of predegraded and non-predegraded poly(lactide) implants: an animal study, J. Mater. Sci.: Mater. Med., 1995, 6, 715–724, DOI:10.1007/BF00134307.
- T. Beslikas, I. Gigis, V. Goulios, J. Christoforides, G. Z. Papageorgiou and D. N. Bikiaris, Crystallization Study and Comparative in Vitro–in Vivo Hydrolysis of PLA Reinforcement Ligament, Int. J. Mol. Sci., 2011, 12, 6597–6618, DOI:10.3390/ijms12106597.
- G. Gorrasi and R. Pantani, Effect of PLA grades and morphologies on hydrolytic degradation at composting temperature: assessment of structural modification and kinetic parameters, Polym. Degrad. Stab., 2013, 98, 1006–1014, DOI:10.1016/j.polymdegradstab.2013.02.005.
- S. Hinderer, M. Schesny, A. Bayrak, B. Ibold, M. Hampel, T. Walles, U. A. Stock, M. Seifert and K. Schenke-Layland, Engineering of fibrillar decorin matrices for a tissue-engineered trachea, Biomaterials, 2012, 33, 5259–5266, DOI:10.1016/j.biomaterials.2012.03.075.
- M. Pudlas, S. Koch, C. Bolwien, S. Thude, N. Jenne, T. Hirth, H. Walles and K. Schenke-Layland, Raman spectroscopy: a non invasive analysis tool for the discrimination of human skin cells, Tissue Eng., Part C, 2011, 17, 1027–1040 CrossRef PubMed.
- Q. Tan, S. Li, J. Ren and C. Chen, Fabrication of Porous Scaffolds with a Controllable Microstructure and Mechanical Properties by Porogen Fusion Technique, Int. J. Mol. Sci., 2011, 12, 890–904, DOI:10.3390/ijms12020890.
- J. Guan, M. S. Sacks, E. J. Beckman and W. R. Wagner, Biodegradable poly(ether ester urethane)urea elastomers based on poly(ether ester) triblock copolymers and putrescine: synthesis, characterization and cytocompatibility, Biomaterials, 2004, 25, 85–96, DOI:10.1016/S0142-9612(03)00476-9.
- Y. Hong, J. Guan, K. L. Fujimoto, R. Hashizume, A. L. Pelinescu and W. R. Wagner, Tailoring the degradation kinetics of poly(ester carbonate urethane)urea thermoplastic elastomers for tissue engineering scaffolds, Biomaterials, 2010, 31, 4249–4258, DOI:10.1016/j.biomaterials.2010.02.005.
- S. Hinderer, J. Seifert, M. Votteler, N. Shen, J. Rheinlaender, T. E. Schäffer and K. Schenke-Layland, Engineering of a bio-functionalized hybrid off-the-shelf heart valve, Biomaterials, 2014, 35, 2130–2139, DOI:10.1016/j.biomaterials.2013.10.080.
- E. Brauchle, H. Johannsen, S. Nolan, S. Thude and K. Schenke-Layland, Design and analysis of a squamous cell carcinoma in vitro model system, Biomaterials, 2013, 34, 7401–7407, DOI:10.1016/j.biomaterials.2013.06.016.
- P. H. Lakner, M. G. Monaghan, Y. Möller, M. A. Olayioye and K. Schenke-Layland, Applying phasor approach analysis of multiphoton FLIM measurements to probe the metabolic activity of three-dimensional in vitro cell culture models, Sci. Rep., 2017, 7, 42730, DOI:10.1038/srep42730.
- S. Lin, R. Wu, H. Tan, W. Lo, W. Lin, T. Young, C. Hsu, J. Chen, S. Jee and C. Dong, Evaluating cutaneous photoaging by use of multiphoton fluorescence and second-harmonic generation microscopy, Opt. Lett., 2005, 30, 2275–2277, DOI:10.1364/OL.30.002275.
- T. G. Scott, R. D. Spencer, N. J. Leonard and G. Weber, Synthetic spectroscopic models related to coenzymes and base pairs. V. Emission properties of NADH. Studies of fluorescence lifetimes and quantum efficiencies of NADH, AcPyADH, [reduced acetylpyridineadenine dinucleotide] and simplified synthetic models, J. Am. Chem. Soc., 1970, 92, 687–695, DOI:10.1021/ja00706a043.
- A. K. Jaiswal, S. S. Kadam, V. P. Soni and J. R. Bellare, Improved functionalization of electrospun PLLA/gelatin scaffold by alternate soaking method for bone tissue engineering, Appl. Surf. Sci., 2013, 268, 477–488, DOI:10.1016/j.apsusc.2012.12.152.
- S. M. Moon, H. Uyama, S. Inoue and Y. Tabata, Fabrication of Non-woven Mats of Gelatin/Poly(L-lactic acid) Composites by Electrospinning and Their Application for Scaffold of Cell Proliferation, Chem. Lett., 2006, 35, 564–565, DOI:10.1246/cl.2006.564.
- K. An, H. Liu, S. Guo, D. Kumar and Q. Wang, Preparation of fish gelatin and fish gelatin/poly(l-lactide) nanofibers by electrospinning, Int. J. Biol. Macromol., 2010, 47, 380–388, DOI:10.1016/j.ijbiomac.2010.06.002.
-
Z. Li and C. Wang, Effects of Working Parameters on Electrospinning, One-Dimensional nanostructures, SpringerBriefs in Materials, 2013, pp. 15–28, DOI:10.1007/978-3-642-36427-3_2.
- N. Nagiah, R. Johnson, R. Anderson, W. Elliott and W. Tan, Highly Compliant Vascular Grafts with Gelatin-Sheathed Coaxially Structured Nanofibers, Langmuir, 2015, 31, 12993–13002, DOI:10.1021/acs.langmuir.5b03177.
- H. W. Kim, H. S. Yu and H. H. Lee, Nanofibrous matrices of poly(lactic acid) and gelatin polymeric blends for the improvement of cellular responses, J. Biomed. Mater. Res., Part A, 2008, 87, 25–32, DOI:10.1002/jbm.a.31677.
- G. Zhu,
et al., Raman spectra of amino acids and their aqueous solutions, Spectrochim. Acta, Part A, 2011, 78, 1187–1195 CrossRef PubMed.
- H. F. Shurvell,
et al., Raman spectra of L(+)-glutamic acid and related compounds, J. Raman Spectrosc., 1989, 20, 163–168 CrossRef.
- J. T. Navarette,
et al., IR and Raman spectra of L-aspartic acid and isotopic derivatives, Biopolymers, 1994, 34, 1065–1077 CrossRef.
- G. Kister,
et al., Effects of morphology, conformation and configuration on the IR and Raman spectra of various poly(lactic acid)s, Polymer, 1998, 39, 267–273 CrossRef.
- D. Qin and R. Kean, Crystallinity Determination of Polylactide by FT-Raman Spectrometry, Appl. Spectrosc., 1998, 52, 488–495 CrossRef.
- C. Vogt,
et al., Degradation of poly(L-lactic acid) coating on permanent coronary metal stent investigated ex vivo by micro Raman spectroscopy, J. Raman Spectrosc., 2017, 48, 711–719, DOI:10.1002/jrs.5111.
- G. Piccirillo, B. Bochicchio, A. Pepe, K. Schenke-Layland and S. Hinderer, Electrospun poly-L-lactide scaffold for the controlled and targeted delivery of a synthetically obtained Diclofenac prodrug to treat actinic keratosis, Acta Biomater., 2017, 52, 187–196, DOI:10.1016/j.actbio.2016.11.002.
- A. Bigi, S. Panzavolta and K. Rubini, Relationship between triple-helix content and mechanical properties of gelatin films, Biomaterials, 2004, 25, 5675–5680, DOI:10.1016/j.biomaterials.2004.01.033.
- J. Y. Lai, D. H. K. Ma, M. H. Lai, Y. T. Li and R. J. Chang,
et al., Characterization of Cross-Linked Porous Gelatin Carriers and Their Interaction with Corneal Endothelium: Biopolymer Concentration Effect, PLoS One, 2013, 8, e54058, DOI:10.1371/journal.pone.0054058.
- T. H. Nguyen and B. T. Lee, The effect of cross-linking on the microstructure, mechanical properties and biocompatibility of electrospun polycaprolactone–gelatin/PLGA–gelatin/PLGA–chitosan hybrid composite, Sci. Technol. Adv. Mater., 2012, 13, 035002, DOI:10.1088/1468-6996/13/3/035002.
- T. Nguyen and B. Lee, Fabrication and characterization of cross-linked gelatin electrospun nano-fibers, J. Biomed. Sci. Eng., 2010, 3, 1117–1124, DOI:10.4236/jbise.2010.312145.
- J. F. Pan, N. H. Liu, H. Sun and F. Xu, Preparation and Characterization of Electrospun PLCL/Poloxamer Nanofibers and Dextran/Gelatin Hydrogels for Skin Tissue Engineering, PLoS One, 2014, 9, e112885, DOI:10.1371/journal.pone.0112885.
- Z. X. Meng, Y. S. Wang, C. Ma, W. Zheng, L. Li and Y. F. Zheng, Electrospinning of PLGA/gelatin randomly-oriented and aligned nanofibers as potential scaffold in tissue engineering, Mater. Sci. Eng., C, 2010, 30, 1204–1210, DOI:10.1016/j.msec.2010.06.018.
- M. Kharaziha, M. Nikkhah, S. Shin, N. Annabi, N. Masoumi, A. K. Gaharwar, G. Camci-Unal and A. Khademhosseini, PGS:Gelatin nanofibrous scaffolds with tunable mechanical and structural properties for engineering cardiac tissues, Biomaterials, 2013, 34, 6355–6366, DOI:10.1016/j.biomaterials.2013.04.045.
- Y. Zhang, H. Ouyang, C. T. Lim, S. Ramakrishna and Z. M. Huang, Electrospinning of gelatin fibers and gelatin/PCL composite fibrous scaffolds, J. Biomed. Mater. Res., Part B, 2005, 72, 156–165, DOI:10.1002/jbm.b.30128.
- X. Jing, M. R. Salick, T. Cordie, H. Mi, X. Peng and L. Turng, Electrospinning Homogeneous Nanofibrous Poly(propylene carbonate)/Gelatin Composite Scaffolds for Tissue Engineering, Ind. Eng. Chem. Res., 2014, 53, 9391–9400, DOI:10.1021/ie500762z.
- K. König, K. Schenke-Layland, I. Riemann and U. A. Stock, Multiphoton autofluorescence imaging of intratissue elastic fibers, Biomaterials, 2005, 26, 495–500, DOI:10.1016/j.biomaterials.2004.02.059.
- K. Schenke-Layland, Non-invasive multiphoton imaging of extracellular matrix structures, J. Biophotonics, 2008, 1, 451–462, DOI:10.1002/jbio.200810045.
- M. Vielreicher, S. Schürmann, R. Detsch, M. A. Schmidt, A. Buttgereit, A. Boccaccini and O. Friedrich, Taking a deep look: modern microscopy technologies to optimize the design and functionality of biocompatible scaffolds for tissue engineering in regenerative medicine, J. R. Soc., Interface, 2013, 10, 1–21, DOI:10.1098/rsif.2013.0263.
- Y. Sun, H. Y. Tan, S. J. Lin, H. S. Lee, T. Y. Lin, S. H. Jee, T. H. Young, W. Lo, W. L. Chen and C. Y. Dong, Imaging tissue engineering scaffolds using multiphoton microscopy, Microsc. Res. Tech., 2008, 71, 140–145, DOI:10.1002/jemt.20537.
- D. Yova, V. Hovhannisyan and T. Theodossiou, Photochemical effects and hypericin photosensitized processes in collagen, J. Biomed. Opt., 2001, 6, 52–57, DOI:10.1117/1.1331559.
- T. S. Blacker, Z. F. Mann, J. E. Gale, M. Ziegler, A. J. Bain, G. Szabadkai and M. R. Duchen, Separating NADH and NADPH fluorescence in live cells and tissues using FLIM, Nat. Commun., 2014, 5, 3936, DOI:10.1038/ncomms4936.
- S. Seidenari, F. Arginelli, C. Dunsby, P. M. W. French and K. König,
et al., Multiphoton Laser Tomography and Fluorescence Lifetime Imaging of Melanoma: Morphologic Features and Quantitative Data for Sensitive and Specific Non-Invasive Diagnostics, PLoS One, 2013, 8, e70682, DOI:10.1371/journal.pone.0070682.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb02026d |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Brigida
Bochicchio
a,
Brigida
Bochicchio