
Self-immolative polymers with potent and selective antibacterial activity by hydrophilic side chain grafting†

Abstract
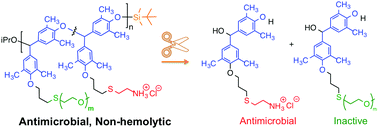
We report the first example of a self-immolative polymer that exerts potent antibacterial activity combined with relatively low hemolytic toxicity. In particular, self-immolative poly(benzyl ether)s bearing pendant cationic ammonium groups and grafted poly(ethylene glycol) chains in their side chains were prepared via post-polymerization thiol–ene chemistry. These functional polymers undergo sensitive and specific triggered depolymerization into small molecules upon exposure to a designed stimulus (in this example, fluoride ions cleave a silyl ether end cap). The molar composition of the resulting statistical copolymers varied from 0 to 100% PEG side chains. The average molar mass of the pendant PEG chains was either 800 or 2000 g mol−1. The antibacterial and hemolytic activities were evaluated as a function of copolymer composition. Strong bactericidal activity (low μg mL−1 MBC) was retained in the copolymers containing 25–50% PEG-800, whereas hemolytic toxicity monotonically decreased (up to HC50 >1000 μg mL−1) with increasing PEG content. PEG-2000 was far less effective; both the MBC and HC50 decreased to a comparable extent with increasing PEGylation. Overall, the best cell type selectivity index (HC50/MBC ∼ 28) was obtained for the copolymer containing ∼50% cysteamine and ∼50% PEG-800 side chains, as compared to the cationic homopolymer (HC50/MBC < 1). Thus, the systematic tuning of the PEG graft density and chain length effectively enhances the cell-type selectivity of these self-immolative polymers by orders of magnitude.
- This article is part of the themed collection: Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

