
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-fouling, mixed-charge poly-L-lysine polymers with anionic oligopeptide side-chains†
Noemi
Bellassai
 abc,
Almudena
Marti
c,
Giuseppe
Spoto
*bd and
Jurriaan
Huskens
*c
abc,
Almudena
Marti
c,
Giuseppe
Spoto
*bd and
Jurriaan
Huskens
*c
aConsorzio Interuniversitario di Ricerca in Chimica dei Metalli nei Sistemi Biologici, c/o Dipartimento di Scienze Chimiche, Università degli Studi di Catania, Catania, Italy
bDipartimento di Scienze Chimiche, Università degli Studi di Catania, Viale Andrea Doria 6, 95125 Catania, Italy. E-mail: spotog@unict.it
cMolecular NanoFabrication Group, MESA+Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: j.huskens@utwente.nl
dConsorzio Interuniversitario Istituto Nazionale Biostrutture e Biosistemi, c/o Dipartimento di Scienze Chimiche, Università degli Studi di Catania, Catania, Italy
First published on 5th November 2018
Abstract
Biosensors and biomedical devices require antifouling surfaces to prevent the non-specific adhesion of proteins or cells, for example, when aiming to detect circulating cancer biomarkers in complex natural media (e.g., in blood plasma or serum). A mixed-charge polymer was prepared by the coupling of a cationic polyelectrolyte and an anionic oligopeptide through a modified “grafting-to” method. The poly-L-lysine (PLL) backbone was modified with different percentages (y%) of maleimide–NHS ester chains (PLL-mal(y%), from 13% to 26%), to produce cationic polymers with specific grafting densities, obtaining a mixed-charge polymer. The anionic oligopeptide structure (CEEEEE) included one cysteine (C) and five glutamic acid (E) units, which were attached to the PLL-mal(y%) polymers, preadsorbed on gold substrates, through the thiol–maleimide Michael-type addition. Contact angle and PM-IRRAS data confirmed monolayer formation of the modified PLLs. Antifouling properties of peptide–PLL surfaces were assessed in adsorption studies using quartz crystal microbalance with dissipation (QCM-D) and surface plasmon resonance imaging (SPRI) techniques. PLL-mal(26%)-CEEEEE showed the best antifouling performance in single-protein solutions, and the nonspecific adsorption of proteins was 46 ng cm−2 using diluted human plasma samples. The new PLL-mal(26%)-CEEEEE polymer offers a prominent low-fouling activity in complex media, with rapid and simple procedures for the synthesis and functionalization of the surface compared to conventional non-fouling materials.
Introduction
The study of protein–inorganic surface interactions is crucial to the rational design of new tools for biomaterials science, biosensing, nanobiotechnology and nanomedicine. Such interactions are responsible for the biocompatibility of materials used in tissue engineering,1,2 biosensors,3,4 and blood-contacting devices.5,6Nonspecific adsorption of proteins can affect surface properties of biomaterials and trigger the degradation of devices operating in contact with biological fluids. When a solid comes in contact with a body fluid such as blood, plasma or serum, a layer of proteins is formed on the surface of the solid material within a time frame of seconds to minutes. For sensors, this may cause functional device interference, possibly preventing the detection of biological targets available at low concentrations in complex media.7–9
Physicochemical properties of surfaces (e.g. topography, electrostatic potential, surface energy), and environmental conditions (e.g. pH, ionic strength and temperature) play an important role in triggering protein–surface interactions.10–12 Proteins show higher affinity for hydrophobic surfaces than hydrophilic ones,10,13 and for rough substrates compared to flat substrates.14
Biomaterial surfaces may be chemically treated to form antifouling layers capable of reducing or, ideally eliminating, fouling.7,9,15 Hydrophilic and bioinert polymers are commonly used to obtain protein-resistant surfaces.16 The hydration layer coupled with the polymeric surface layers acts as a barrier to prevent protein fouling. Moreover, flexibility of the polymer chains reduces the nonspecific adsorption of protein via steric exclusion mechanisms.17
Antifouling materials are classified according to the chemical composition of the hydrophilic polymer. Common materials are based on poly or oligo(ethylene glycol) (PEG/OEG), polysaccharides, zwitterionic compounds such as phosphorylcholine-based derivatives,18,19 betaines, and polypeptides/peptoids. Nonionic OEG and PEG are used extensively to fabricate antifouling coatings exploiting the flexibility and mobility of PEG chains, and the formation of a highly hydrated PEG layer on the solid surface to provide resistance to nonspecific adsorption.20,21 However, low surface densities22 and susceptibility to oxidative damage limit the antifouling capabilities of PEG-based materials in long-term applications.
Zwitterionic materials have emerged as promising candidates for advanced antifouling/biocompatible materials, because of their high hydration capacity and electroneutrality.23,24 Zwitterionic polymers exhibit balanced anionic/cationic groups on their molecular chains, which make them highly hydrophilic and antifouling, while maintaining overall charge neutrality.22,23 In detail, the interfacial water structure in the zwitterionic polymer plays an important role in determining the antifouling performance of the surface, because these polymers are able to strongly bind water molecules and to assemble a hydration film thanks to the balanced network of positive and negative charges inside the polymer structure. Therefore, the strong interfacial hydration or the domination of strongly hydrogen-bonded water at the interface leads to excellent antifouling performance of zwitterionic materials.21,25 Moreover, zwitterionic polymers show higher stability to oxidation than PEG polymers,26 making them a valid alternative to the widely used PEG-based materials.27
While the zwitterionic polymers exhibit promising antifouling capacities, the synthesis of these polymers is difficult and costly. Besides, a critical factor determining nonfouling properties of polyzwitterionic materials is to control both uniformity of charge distribution and charge neutrality of two oppositely charged moieties on the surface. Such factors can be controlled either by using zwitterionic units,28–32 or, more easily, by mixing positively and negatively charged moieties in mixed-charge self-assembled monolayers (SAMs),28,33 polymer coatings,34 or hydrogels.35
Poly-L-lysine (PLL) is a versatile polymer, composed of positively charged lysine amino acid as a repeat unit, which has attractive biochemical properties, including hydrophilicity, excellent biocompatibility and an acceptable degree of biodegradability. Because PLL is positively charged at physiological pH, it can be easily adsorbed on a large variety of negatively charged substrates via electrostatic interactions, including glass,36 metals,37 polymers,38 and metallic oxides.39 Furthermore, PLL polymers can be easily modified with nonionic side-chains (like PEG/OEG), thereby making it an ideal candidate for engineering biomaterial interfaces, such as surface coatings,37,40 drug,41 gene,42 and protein43 delivery platforms, and hydrogel scaffolds.44 The easy functionalization of PLL polymers with different functional groups allows to have a critical control over the biosensing interfaces by creating mixed monolayers with different functionalities.45 The surface modification with PLL polymers grafted with negatively charged side-chains for creating mixed-charge antifouling materials has not yet been performed.
Here, we describe the development of a mixed-charge polymer based on PLL modified with an anionic peptide that is connected via a nonionic OEG spacer, in order to achieve control over the charge distribution of PLL-coated surfaces and thus antifouling character. The PLL backbone was functionalized with different percentages (y%) of maleimide-OEG-NHS ester chains (PLL-mal(y%), from 13% to 26%), to study its effect on the antifouling properties. The anionic oligopeptide CEEEEE, composed of one cysteine (C) and five glutamic acids (E), has a short sequence to limit the thickness of the mixed-charge polymer antifouling coating, and the grafting density can be varied to tune the balance of charged groups at polymer backbone. Upon the adsorption of PLL-mal(y%) polymer on a gold surface, the anionic peptide CEEEEE is attached to the maleimide units through the thiol–maleimide Michael-type addition, thereby creating the antifouling PLL-mal(y%)-CEEEEE polymer on the surface in a two-step simple process. Largely, the structure of the mixed charged polymers proposed here allows for binding water molecules strongly and thus for generating a hydrophilic substrate to prevent the adsorption of biological materials onto the surface.
PLL-mal(y%)-CEEEEE surfaces were characterized by water contact angle and polarization modulation infrared reflection–absorption spectroscopy (PM-IRRAS). Complementary acoustic (quartz crystal microbalance with dissipation, QCM-D) and plasmonic (surface plasmon resonance imaging, SPRI) techniques were employed to monitor the adsorption of bovine serum albumin (BSA), used as standard single-protein solution, and diluted human plasma samples.46
Results and discussion
Positive charges of the PLL backbone play a key role in the immobilization of functionalized PLL polymers on negatively charged surfaces. Modified PLL layers tend to lose their adhesive properties with increasing grafting density of OEG-maleimide chains, and 60% (or more) of free lysine is required for tightly anchoring the positively charged polymer to the activated negatively charged substrates.47 Therefore, only a limited amount of OEG-maleimide chains can be added to PLL without altering the capacity of the whole system to properly immobilize on surfaces. Here, we synthesized PLL-mal(y%) with different percentages of OEG-maleimide (y% ranging from 13% to 26%), to allow coupling with a cysteine-modified peptide by the thiol–maleimide Michael-type addition. To preserve the PLL-mal(y%) capacity to interact with gold, y% values no greater than 26% were considered in this work.The protocol of the synthesis for PLL polymers was adapted from a procedure described elsewhere (Fig. S1, ESI†),47 and the process was carried out under controlled pH conditions to prevent the degradation of the mal moiety (stable until pH 7.5). After the modification with different percentages of mal units, the grafting density of PLL-mal(y%) polymers was calculated using 1H NMR (Fig. S2–S5, ESI†). The anionic peptide CEEEEE was synthesized through the automated solid-phase peptide synthesis (SPPS), and then characterized by HPLC-MS (Fig. S6, ESI†).
Scheme 1 shows a representation of the two-step formation of PLL-mal(y%)-CEEEEE polymers on a gold chip. After the activation of the gold surface by UV-ozone, PLL-mal(y%) polymers were adsorbed on the substrate via electrostatic interactions between the positively charged polymer backbone and the negatively charged surface (Scheme 1, step 1). Thereafter, the anionic peptide CEEEEE was coupled to the mal units of the adsorbed PLL-mal(y%) polymers through the thiol–maleimide Michael-type addition employing the cysteine residue of the CEEEEE sequence (Scheme 1, step 2).
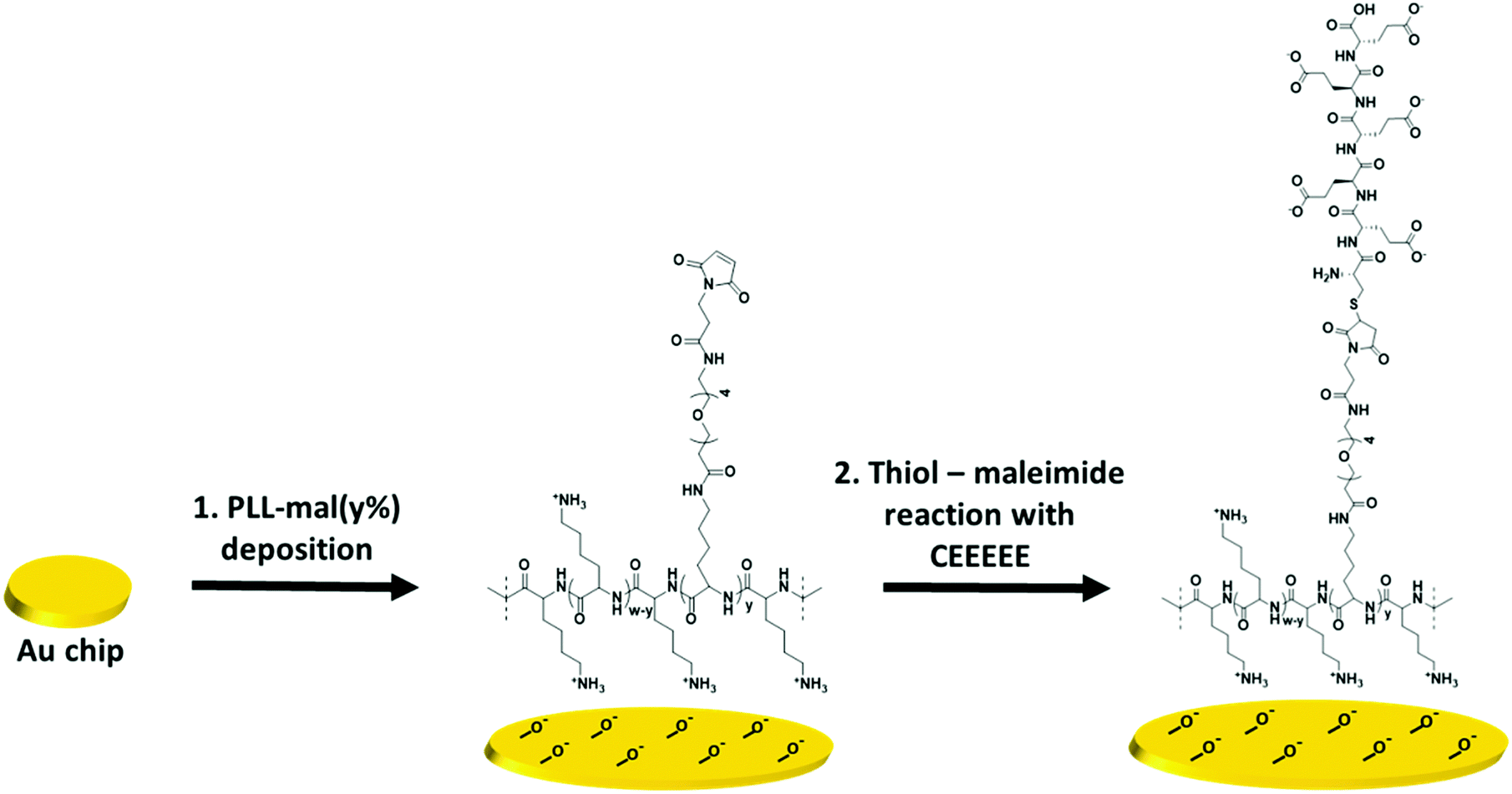 | ||
| Scheme 1 Immobilization of the PLL-mal(y%) polymers on a gold surface (step 1), thiol–maleimide reaction between the anionic peptide CEEEEE and the maleimide units of immobilized PLL-mal(y%) polymers (step 2). | ||
Characterization of PLL-mal(y%)-CEEEEE layers
Water contact angle and PM-IRRAS measurements were used to evaluate the wettability of the substrates and to characterize the PLL-mal(y%)-CEEEEE monolayers deposited on the gold surface. Table 1 shows results from static water contact angle measurements of Au bare, PLL-mal(y%) layers with different percentages of mal units (y%, from 13% to 26%), and PLL-mal(y%)-CEEEEE layers after the coupling reaction of CEEEEE.| Surfaces | (y%) = 26% | (y%) = 22% | (y%) = 13% |
|---|---|---|---|
| θ (°) | θ (°) | θ (°) | |
| Au bare | 86 ± 1 | 86 ± 1 | 86 ± 1 |
| PLL-mal(y%) | 31 ± 1 | 32 ± 1 | 31 ± 1 |
| PLL-mal(y%)-CEEEEE | 55 ± 2 | 53 ± 2 | 50 ± 1 |
Upon adsorption of PLL-mal(y%) on the gold surfaces, the contact angle decreased from 86° to 31° for all PLL-mal polymers tested, irrespective of the degree of mal functionalization (ranging from 13–26%). The low values for the PLL-mal layers indicate a high hydrophilicity of all PLL-mal(y%) surfaces, and the values correspond to values observed for PLL-OEG monolayers before.47 The coupling reaction of CEEEEE to the PLL-mal(y%) layers induced an increase of the contact angle values, reaching 50 to 55°. This increase is attributed to the rise of the grand average of hydropathy (GRAVY) value (−2.5) obtained for the CEEEEE sequence from the GRAVY Calculator,48 compared to the hydropathy index (HI) of lysine residues (−3.9) calculated by the ProtScale software. This difference explains the variation of water contact angle data after the coupling reaction and, consequently, the reduction of the hydrophilicity of the surface (see also ESI†).
Fig. 1 shows representative PM-IRRAS spectra obtained for PLL-mal(26%) deposition on gold before and after the coupling reaction of CEEEEE. The signals in the 1775–1779 cm−1 range are assigned to the symmetric stretch of maleimide carbonyl groups, and the peak at 1712 cm−1 is attributed to the in-phase stretching vibration of the two maleimide carbonyl moieties.49 The disappearance of these groups after the reaction with the peptide confirms the coupling of CEEEEE to the mal units of PLL polymers. The large band at 1654 cm−1 is attributed to the carbonyl stretch of the amide I bonds within the PLL polymer.49 This peak at 1654 cm−1 increased in intensity upon the immobilization of CEEEEE on PLL-mal(y%), by indicating the coupling reaction with the mal moieties as well. In fact, the immobilized CEEEEE adds carboxylate groups to carbonyl moieties already available on the surface after PLL-mal(y%) deposition. The bands at 1524–1560 cm−1 are attributed to the amide II band (CN stretch and NH bend) of PLL-mal(y%) layers, and their peak intensities decreased after the immobilization of CEEEEE peptide, as observed in literature for similar PLL systems functionalized with oligonucleotides.50 Bands in the 1457–1470 cm−1 range are attributed to δ(CH2) scissors deformation, while the band at 1377 cm−1 results from the stretching of (C–N–C) groups. PLL-mal(y%)-CEEEEE polymers with different mal fractions (y%) showed features similar to those here discussed (Fig. S6 and S7, ESI†). The analysis of the PM-IRRAS spectra indicates the successful formation of PLL-mal(y%)-CEEEEE on the gold surface.
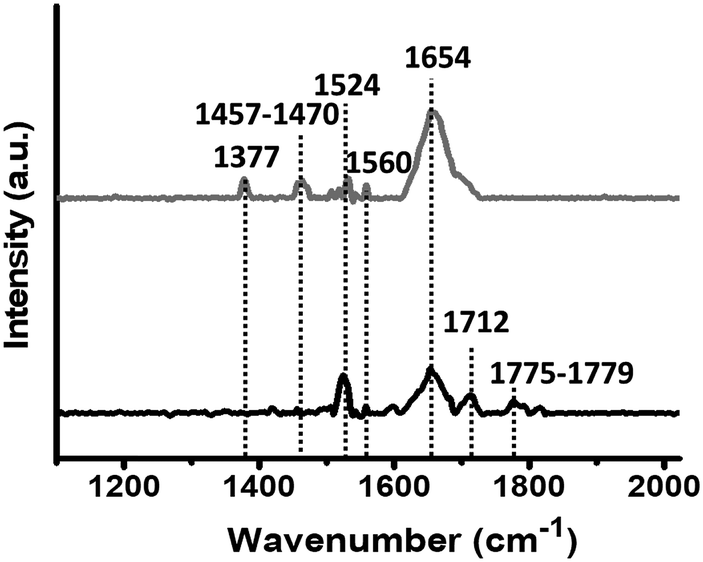 | ||
| Fig. 1 PM-IRRAS spectra of PLL-mal(26%) deposited on a gold chip (black line, bottom), and PLL-mal(26%)-CEEEEE (grey line, top) after the coupling reaction with CEEEEE. | ||
Coupling efficiency of peptide–maleimide unit reaction
Preliminary evaluations for the coupling efficiency of CEEEEE to the PLL-mal(y%) and for the antifouling property of the new PLL-based systems carrying different y% of mal moieties were performed by QCM-D (Table 2), using BSA (1.0 mg mL−1 and 50.0 mg mL−1) as standard single-protein solutions.51 The BSA solution at higher concentration (50.0 mg mL−1) has been utilized as it reflects the average concentration of blood plasma proteins.7,51| (y%) | PLL-mal(y%) | CEEEEE | Mol ratio CEEEEE/mal(y%) | [BSA] 1.0 mg mL−1 | [BSA] 50.0 mg mL−1 |
|---|---|---|---|---|---|
| 26 | 291 | 470 | 1.9 | 32 | 41 |
| 22 | 363 | 648 | 2.3 | 20 | 45 |
| 13 | 406 | 446 | 2.0 | 29 | 49 |
Even though the coupling reaction of CEEEEE to PLL-mal, to give PLL-mal(y%)-CEEEEE, induced an increase of the water contact angle values and, thus, a reduction of the surface hydrophilicity (Table 1), the antifouling behavior is related to the net charge of the peptide, which equals to −5 at physiological pH. In particular, the network of positive and negative charges inside the polymer structure, obtained after the functionalization of the PLL-mal(26%) layer with CEEEEE, may play a synergistic role to the fouling protein resistance.
X-ray photoelectron spectroscopy (XPS) provided important chemical state information regarding PLL-mal(26%) and PLL-mal(26%)-CEEEEE monolayers.
An XPS survey scan of a PLL-mal(26%) layer deposited on gold surface (Fig. S9 and Table S1, ESI†) shows signals attributed to Au, C, O, N and Cl, to be compared with Au, C and O signals detected from a bare gold surface (Fig. S8, ESI†). The presence of such signals, together with the increased intensity of the signals attributed to C and O, confirmed the deposition of the amino acid-based PLL-mal(26%) layer. A survey scan of the deposited PLL-mal(26%)-CEEEEE layer confirmed the presence of the thiolated CEEEEE moiety, as testified by S 2s and S 2p signals (Fig. S10 and Table S1, ESI†).
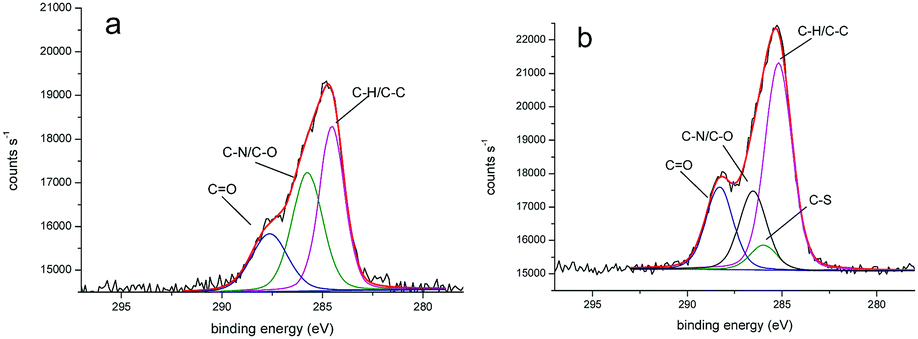
We obtained more detailed information about the deposited layers by evaluating XPS core spectra. In particular, the deconvolution of C 1s region allowed us to identify three different chemical environments producing signals attributed to C–C, C–O/C–N, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moieties and centered at 284.7, 286.0, 287.7 eV for PLL-mal(26%) and 284.9, 286.3, 288.0 eV for PLL-mal(26%)-CEEEEE, respectively (Fig. 2).52,53 An additional band centered at 285.7 eV and attributed to C–S bonds54 was found to contribute to the PLL-mal(26%)-CEEEEE C 1s core spectrum. For the curve-fitting procedure, all C 1s peaks were described using a Gaussian/Lorentzian percent ratio of 80%. The full-width-half-maximum (FWHM) of each component C 1s peaks were set to 1.78 eV for PLL-mal(26%) and 1.70 eV for PLL-mal(26%)-CEEEEE, respectively.
O moieties and centered at 284.7, 286.0, 287.7 eV for PLL-mal(26%) and 284.9, 286.3, 288.0 eV for PLL-mal(26%)-CEEEEE, respectively (Fig. 2).52,53 An additional band centered at 285.7 eV and attributed to C–S bonds54 was found to contribute to the PLL-mal(26%)-CEEEEE C 1s core spectrum. For the curve-fitting procedure, all C 1s peaks were described using a Gaussian/Lorentzian percent ratio of 80%. The full-width-half-maximum (FWHM) of each component C 1s peaks were set to 1.78 eV for PLL-mal(26%) and 1.70 eV for PLL-mal(26%)-CEEEEE, respectively.
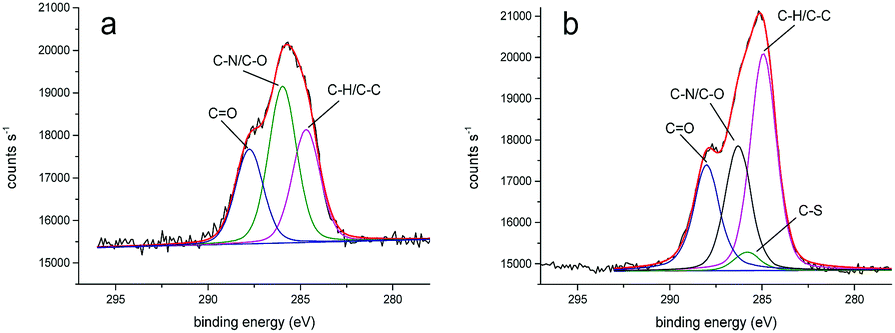 | ||
| Fig. 2 Representative XPS C 1s core scans for PLL-mal(26%) (a) and PLL-mal(26%)-CEEEEE (b) layers deposited on gold. The red line represents the convolution of bands attributed to C–H/C–C, C–S, C–O/C–N, and CO bonds. | ||
The evaluation of the intensity ratio of bands attributed to C–H/C–C, C–N/C–O and CO provided additional evidence of the contribution of CEEEEE to the surface monolayer (Table 3). The increase of (C–H/C–C)/(C–N/C–O) and (C–H/C–C)/CO ratios after the coupling reaction with the peptide, is evidence of the successful immobilization of CEEEEE on modified PLL-mal(26%) surface.
| Band | Chemical state | Position (eV) | Area (%) | Band int. ratio | Band int. ratio | |
|---|---|---|---|---|---|---|
| PLL-mal(26%) | 1 | C–H/C–C | 284.7 | 30.97 | ||
| 2 | C–N/C–O | 286.0 | 43.07 | 0.98 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) :2) |
||
| 3 | CO |
287.7 | 25.95 | 1.17 (1:3) |
||
| PLL-mal(26%)-CEEEEE | 1 | C–H/C–C | 284.9 | 45.23 | ||
| 2 | C–S | 285.7 | 2.97 | |||
| 3 | C–N/C–O | 286.3 | 26.65 | 1.70 (1:3) |
||
| 4 | CO |
288.0 | 25.14 | 1.80 (1:4) |
||
The deconvolution of the N 1s core spectra was obtained by considering one band at 399.4 eV for PLL-mal(26%) and one at 399.7 eV for PLL-mal(26%)-CEEEEE, respectively. The band is attributed to the C–N (amine and amide) bond. After the reaction of the CEEEEE peptide, an increase of the N 1s peak intensity was detected thus suggesting a successful immobilization of the peptide on the PLL-mal(26%) surface (Fig. S11, ESI†).
The study of the S 2p region can provide useful information on chemical state of sulfur in thiol/sulfide compounds reacting with organic or inorganic (e.g. gold) species.55
We found no S 2p signals when analyzing PLL-mal(26%) deposited on a gold substrate. S 2p signals with a doublet structure due to the presence of the S 2p3/2 and S 2p1/2 peaks were instead observed from PLL-mal(26%)-CEEEEE (Fig. 3a). Two S 2p3/2/S 2p1/2 doublets contributed to the detected S 2p signal. The S 2p3/2 component at 162.1 eV is consistent with the sulfur atoms bound to the gold surface as a thiolate species, while S 2p3/2 component at 164.1 eV is attributed to Au-unbound thiols that, in our case, are reasonably attributed to maleimide-bound sulfur atoms. However, it is underlined that an easy discrimination between S–H and S–C S 2p XPS signals is not possible.55 The ratio of the intensity of the unbound/bound S 2p signals allows to conclude that about 35% of sulfur atoms are not directly bound to the gold surface.
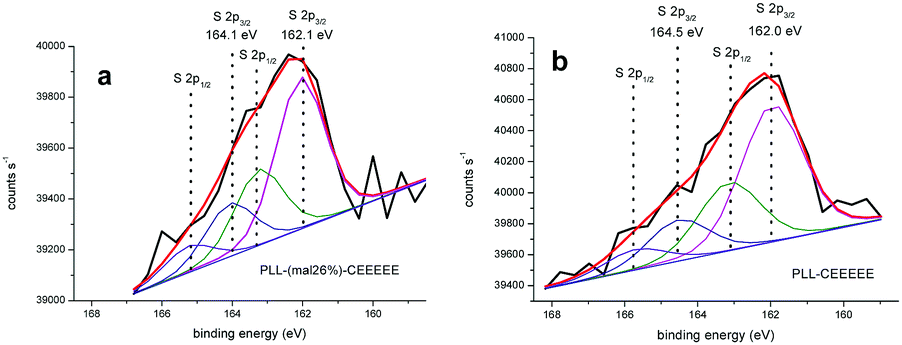 | ||
| Fig. 3 Representative XPS S 2p spectra for (a) PLL-mal(26%)-CEEEEE and (b) PLL-CEEEEE films adsorbed onto gold surfaces. The S 2p peaks were fit using one S 2p doublet with a 2:1 area ratio and a splitting of 1.18 eV. The position of the S 2p3/2 peaks assigned to Au-bound thiolate and Au-unbound/maleimide bound thiol species are shown. The FWHM of S 2p component peaks were set to 1.70 eV and 1.80 eV for PLL-mal(26%)-CEEEEE and PLL-CEEEEE, respectively. Gaussian/Lorentzian percent ratio was fixed to 80% for both XPS analysis. | ||
XPS was also employed to assess the level of immobilization of CEEEEE on the PLL surface without maleimide linkers. The S 2p core scan (Fig. 3b) showed also two S 2p3/2/S 2p1/2 doublets with S 2p3/2 signals at 162.0 and 164.5 eV. The 0.4 eV shift of the signal attributed to Au–unbound sulfur atoms testifies the partial oxidation of the original thiol moiety that may be responsible for the non-specific adsorption of CEEEEE on PLL. The intensity of the signal produced by the partially oxidated thiolated moiety is 30% of the signal produced by Au–bound sulfur atoms.
We also examined C 1s core levels. Deconvolution procedures provided signals at 284.6, 285.9, 287.7 eV for PLL and 285.2, 286.0, 286.5, 288.3 eV for PLL-CEEEEE (Fig. 4a and b). No significant variations in the band position were detected testifying that non-specific adsorption on PLL and specific interaction between the CEEEEE thiol moiety and gold are both present when the maleimide unit is not present in the structure. However, oxidation of the thiol moiety seems to contribute to non-specific absorption of CEEEEE on PLL instead of the specific binding observed when CEEEEE reacts with PLL-mal(26%).
 | ||
| Fig. 4 XPS C 1s signals of PLL (a) and PLL-CEEEEE (b) layers on gold substrates. The red line is the cumulative curve for the deconvoluted peaks, while the black line shows the unmodified data. | ||
Furthermore, the XPS analysis was employed to investigate the maleimide content of the different percentages of linkers (from 13% to 26%) (Table S2, ESI†). As expected, the XPS spectra show the gradual increase of carbon and nitrogen contents from the lowest to the highest percentage of maleimide units, which confirm the different degrees of PLL polymer functionalization.
QCM-D was used to assess film formation, peptide coupling, and protein adsorption. All the analyses of QCM-D measurements (Δf and ΔD) revealed that the ratio ΔD/(ΔfN/N, where N is the overtone) remained below 0.4 × 10−6 Hz−1 and, therefore, we considered the Sauerbrey equation valid to estimate the adsorbed mass of PLL-mal(y%), peptide and proteins, assuming the film as rigid.56
Upon the use of QCM-D to assess the coupling efficiency of CEEEEE and PLL-mal(y%), it was observed (Table 2) that the adsorbed masses of peptide were approx. twice those expected to get saturation of the mal units. These differences can possibly be attributed to stronger hydration of CEEEEE compared to PLL, which could arise from the tight adsorption of PLL to the substrate while the peptide chains stand out from the surface. This would mean that in particular the areal masses of the peptide are overestimated, and thus also the peptide/mal ratios. Yet, a minor contribution from insertion of the peptide directly onto the gold surface cannot be excluded.
When assuming that each peptide has 5 negative charges at pH 7.4, charge neutrality is reached when a 17% mal-functionalized PLL (1 neutral mal-functionalized unit per 5 positively charged unfunctionalized lysine units) is fully reacted with the peptide. With the results presented above, it can be estimated that the resulting layers are ranging, for increasing mal fractions, from close-to charge neutral to negatively charged. Over-charging occurs also normally for polyelectrolytes and it depends on the conditions during coupling reaction. In fact, background salt screens the surface charges making it continuously accessible for the peptide, even when the surface is already overall negatively charged. Moreover, it was observed that the highest percentage of mal units (26%) provides better antifouling performances than the other PLL-mal(y%)-CEEEEE polymers, with the lowest surface coverage value (41 ng cm−2) for 50.0 mg mL−1 of BSA. We believe that the highest degree of mal units provides more binding sites available for the coupling reaction with CEEEEE, thus leading to more negative charges on the surface and improving the resistance to the adsorption of negatively charged proteins at physiological pH.57
SPRI and QCM-D were used for the comparison of coupling efficiency for the reaction of CEEEEE to the PLL-mal(y%) layer in real time. Fig. 5 shows representative SPRI and QCM-D curves for the adsorption of PLL-mal(26%) (Fig. 5a and b) on gold substrate and the coupling reaction of CEEEEE to mal moieties of PLL-mal(26%) layer (Fig. 5c and d).
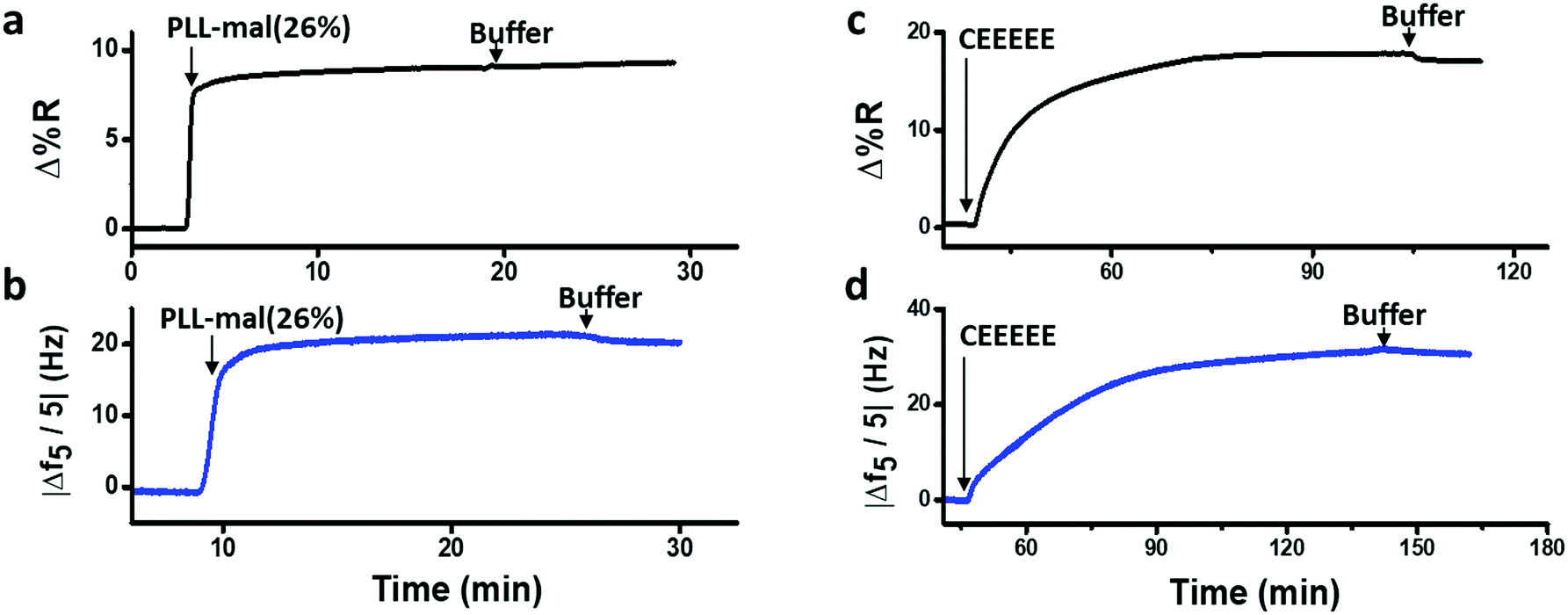 | ||
| Fig. 5 Representative SPRI (a and c) and QCM-D (b and d) adsorption curves of the immobilization of PLL-mal(26%) on gold (a and b), and of the subsequent coupling of CEEEEE to the maleimide units (c and d). | ||
Both SPRI and QCM-D data confirmed that the adsorption of PLL-mal(26%) on gold proceeds with fast kinetics, reaching saturation within a few minutes, and providing a stable surface layer as witnessed by the absence of desorption upon switching to buffer (Fig. 5a and b). The coupling reaction between CEEEEE and the PLL-mal(26%) layer adsorbed on gold appeared to go slower (Fig. 5c and d), as both techniques indicate saturation taking at least 30 min. The areal masses of the absorbed layers and the molar ratio between CEEEEE and PLL-mal(26%) were also calculated considering the SPRI responses after the immobilization of the studied systems (see Table S3, ESI†). In particular, relevant differences were calculated for the areal mass of the PLL-mal(26%) layer by SPRI (138 ng cm−2, Fig. 5a and Table S4, ESI†) and QCM-D (291 ng cm−2Fig. 5b and Table 2), and also different values of the areal mass after the adsorption of CEEEEE were measured by SPRI (282 ng cm−2, Fig. 5c and Table S3, ESI†) and QCM-D (470 ng cm−2, Fig. 5d and Table 2). Similar results for the peptide–mal molar ratio were obtained by both techniques (2.4 by SPRI, Table S3, ESI† and 1.9 by QCM-D, Table 2).
For the mass density measurements, significant variations were observed between SPRI and QCM-D results, where the PLL-mal(26%) and CEEEEE layers are heavily hydrated in QCM-D, nearly twice as much when compared with the corresponding SPRI data. While the hydration of PLL-mal(26%) and CEEEEE may be differently weighed in QCM-D experiments, due to the bound water molecules co-measured with the PLL polymer and, especially, with the peptide, similar molar ratios of CEEEEE to PLL-mal(26%) were obtained by SPRI and QCM-D, confirming the same coupling efficiency of the reaction obtained by two different techniques.
Antifouling property in BSA solution
Subsequently, the antifouling activities for PLL-mal(26%)-CEEEEE were investigated by both QCM-D and SPRI, using the same BSA solutions. Fig. 6 shows representative SPRI (Fig. 6a) and QCM-D (Fig. 6b) responses measured during the adsorption of BSA (1.0 mg mL−1 and 50.0 mg mL−1) on PLL-mal(26%)-CEEEEE.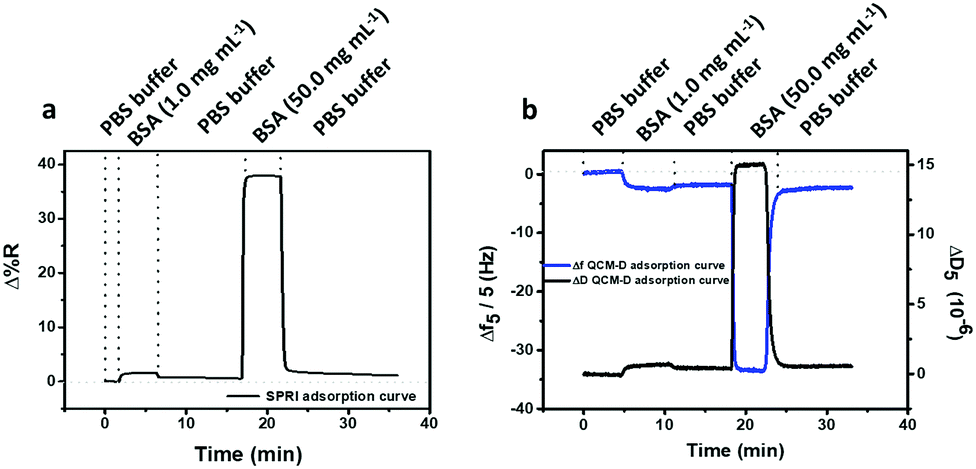 | ||
| Fig. 6 BSA adsorption on PLL-mal(26%)-CEEEEE measured by (a) SPRI and (b) QCM-D. Frequency (blue line) and energy dissipation (black line) shifts corresponding to BSA adsorption are observed in QCM-D measurement (b). | ||
Surface coverages were similarly quantified by SPRI when a 1.0 mg mL−1 and 50.0 mg mL−1 solutions were put in contact with the PLL-mal(26%)-CEEEEE antifouling layer for 5 min, respectively (Table S3, ESI†). In contrast, calculations based on QCM-D experiments produced a higher surface coverage (41 ng cm−2) when 26% of maleimide units was considered (Table 2). It has been already reported that QCM-D data are significantly affected by the contribution (70–90%) of bound water molecules present in the structure of adsorbed molecules, in particular proteins, such as BSA.46 On the basis of the experimental data, it is evident that water contributes to the QCM frequency change to a higher extent.
Only for the PLL-mal(26%)-CEEEEE layer, both QCM and SPR were performed, for the other mal fractions only QCM-D was used (see Table 2). The comparison of layers with different mal fractions shows that the best antifouling properties were obtained for the highest degree of peptide functionalization.
Compared with the polymer systems of antifouling coatings in SPRI experiments, such as polysaccharide-based materials and zwitterionic compounds based materials where the protein adsorption levels were more than 6 ng cm−2 using lower concentrations of BSA, such as 0.1 mg mL−1 and 1.0 mg mL−1,58–60 the functionalized PLL-mal(26%)-CEEEEE layer exhibits an excellent resistance to fouling from highly concentrated single-protein solutions, as revealed by SPRI data. A further non-fouling investigation was performed on silicon oxide surfaces covered with PLL-mal(26%)-CEEEEE, by QCM-D experiments (Fig. S12 and Table S4, ESI†). Similar antifouling performance was achieved when PLL-mal(26%)-CEEEEE layer was immobilized on SiO2 (55 ng cm−2) as compared to modified PLL on a gold surface (41 ng cm−2), by confirming the antifouling character of the layer on both surfaces in BSA solutions.
Antifouling property in human plasma sample
In order to assess the antifouling activity of the PLL-mal(26%)-CEEEEE layer in real and complex biological samples, diluted human blood plasma samples (5%, 10% and 33% in PBS) were tested (Fig. 7) and, thereby, the final adsorbed protein mass was calculated by SPRI and QCM-D (Table 4).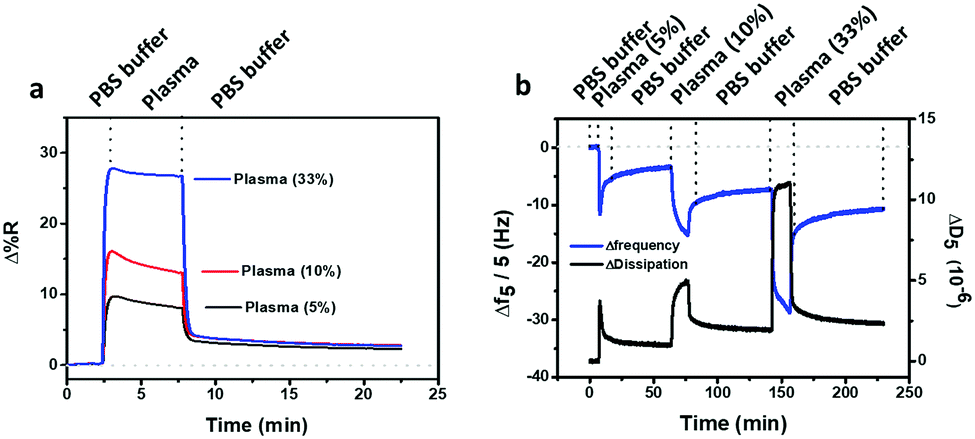 | ||
| Fig. 7 (a) SPRI and (b) QCM-D antifouling tests using diluted human plasma samples and PLL-mal(26%)-CEEEEE. Frequency (blue line) and energy dissipation (black line) shifts corresponding to the adsorption of protein from diluted human plasma samples are observed in QCM-D measurement (b). | ||
| Human plasma in PBS (%) | SPRI (ng cm−2) | QCM-D (ng cm−2) |
|---|---|---|
| 5 | 39 | 62 |
| 10 | 48 | 130 |
| 33 | 46 | 190 |
As reported in Table 4, it was revealed that the resistance to fouling from complex media does not reflect the surface resistance to single-protein BSA solutions. Human plasma comprises a mixture of plasma proteins, being lysozyme (MW about 14 kDa), albumin (MW about 66 kDa), immunoglobulins (MW about 150 kDa) and fibrinogen (MW about 340 kDa) the most abundant.46,61 The hydration shell of bound water molecules, the concentration and the molecular size of the plasma proteins have a relevant effect on the adsorption phenomenon and, then, on the mass of adsorbed protein detected by QCM-D (Table 4). In general, small proteins, such as lysozyme, are able to penetrate the antifouling polymer and adsorb directly on the substrate. This process is called primary adsorption, or ternary adsorption when the small proteins are within the layer. Both adsorption mechanisms strongly influence the QCM resonance frequency shifts, where the smallest proteins can be better detected than the largest ones.46 The adsorption “on top” of the layer, termed secondary adsorption, is more likely to arise for large plasma protein, such as fibrinogen. Depending on the thickness of the antifouling polymer, the secondary adsorption process affect QCM-D results and the absolute amount of protein absorbed may be underestimated when this adsorption process on top of the layer occurred.46 Here, the thickness of the antifouling layers (few nm) allows the small proteins to penetrate within the layer, by strongly affecting the overestimation of the mass density for proteins adsorbed.
The values of adsorbed mass, equal to 46 ng cm−2 using 33% human plasma detected by SPRI (Table 4), show a significant non-fouling activity of PLL-mal(26%)-CEEEEE layer in real human plasma samples, especially if compared to PEG/OEG-based materials and other hydrophilic antifouling materials where the protein adsorption levels in complex media were more than 60 ng cm−2 in SPR measurements.62–65 Moreover, the antifouling test was carried out for PLL-mal(26%)-CEEEEE deposited on a silicon oxide surface, thus confirming the antifouling property in diluted human plasma samples comparable to the previous results obtained for the gold surface (Fig. S13 and Table S5, ESI†).
To assess the contribution to the antifouling properties of the coupling reaction of CEEEEE with PLL-mal(26%), the antifouling test was replicated using 33% diluted human plasma on PLL-mal(26%) surface, in the absence of the peptide CEEEEE. The large amount of adsorbed protein (74 ± 3 ng cm−2) measured by SPRI is attributed to electrostatic interactions between the positively charged surface and the oppositely charged protein residues. Hence, the increase of the nonspecific protein adsorption of PLL-mal(26%) confirmed that the CEEEEE plays a crucial role for the antifouling property of PLL-mal(26%)-CEEEEE layer.
Experimental
Synthesis of oligopeptide CEEEEE
As the solvents for the automated solid-phase peptide synthesis (SPPS) using Multisyntech GmbH instrument, 1-Hydroxybenzotriazole hydrate (HOBt), to dissolve the amino acid solutions, 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), N,N-diisopropylethylamine (DIPEA), N-methyl-2-pyrrolidone (NMP), piperidine, dichloromethane (DCM) and methanol were purchased from Sigma-Aldrich. For the manual cleavage and precipitation trifluoroacetic acid (TFA), triisopropylsilane (TIPS), 1,2-ethanedithiol (EDT) and diethyl ether solvents were obtained from Sigma-Aldrich (The Netherlands). Milli-Q water with a resistivity >18 MΩ cm was used in all experiments.
The oligopeptide Cys-Glu-Glu-Glu-Glu-Glu (or CEEEEE) was synthesized by SPPS using the Fmoc-Glu(OtBu)-Wang resin (Sigma-Aldrich, The Netherlands). The linker attached to the polystyrene core is a 4-hydroxybenzyl alcohol moiety, modified with glutamate (OtBu)-OH used as the first amino acid.
The protocol consisted of five cycles of synthesis using single coupling mode for the first cycle, then the double coupling mode was actuated to ensure the complete activation of carboxyl groups, indispensable for speeding up the reaction. In every cycle, one glutamic acid was attached to the peptide sequence and the last amino acid was a cysteine residue. The active ester group for the carboxyl groups was introduced as a phosphonium salt of a non-nucleophilic anion (hexafluorophosphate) using HBTU/HOBt coupling activation and DIPEA/NMP solution. Thoroughly, four vials for the peptide were prepared by weighing 50 mg of Fmoc-Glu(OtBu)-Wang resin per vial. Thereafter, the resin was filled with NMP for the swelling phase for 2 h. Fmoc-Cys(Trt)-OH (0.29 M) and Fmoc-Glu(OtBu)-OH (0.29 M) were dissolved in 0.30 M HOBt/NMP solution, and 0.26 M of HBTU solution in NMP solvent for the coupling activation of the amino acids. Before the addition of the single amino acids, during each cycle of synthesis 20% of piperidine in NMP was used for removing the Fmoc protection group of the last amino acid in the sequence. This cleavage step was replicated for five cycles of synthesis. The peptide was rinsed with NMP, DCM and methanol (three cycles for three hours); then, the product was dried overnight under vacuum.
The cocktail cleavage for CEEEEE was composed with 94.75% TFA/2.5% TIPS/0.25% EDT/2.5% Milli-Q water. During the cleavage, the peptide was gently stirred for 4 h to avoid breaking the resin. Subsequently, the resin was rinsed with TFA, and all of the organic scavengers were removed using the rotavapor.
For the precipitation, diethyl ether was added to the peptide, and the sample was collected after three/four centrifugation steps. Afterwards, the precipitate was dissolved and treated with the lyophilization, after that the lyophilized product stored under Argon flow to prevent the oxidation of the peptide.
The purification of CEEEEE was performed using high performance liquid chromatography (HPLC) on Water (2535) setup equipped with analytical and preparative XBridge C18 columns. The peptide solution was dissolved in H2O with 0.1% TFA and purified by gradient elution method (linear gradient from H2O (99%) to acetonitrile (ACN, 100%)). The retention time of the oligopeptide was 10 min. The oligopeptide was characterized by mass spectrometry (Fig. S5, ESI†).
Synthesis of poly-L-lysine-g-maleimide(y%) (PLL-mal(y%))
Phosphate buffered saline tablets (PBS, pH 7.4), and poly-L-lysine·HBr (PLL·HBr) (15–30 kDa) were purchased from Sigma-Aldrich. (NHS)-tetra(ethylene glycol)-maleimide (NHS-(OEG4)-mal) and Zeba™ Spin Desalting Columns (7 kD MWCO, 5 mL) were purchased from Thermo Fischer Scientific. 1H-NMR and 13C-NMR spectra were recorded on a Bruker 400 MHz spectrometer. Chemical shifts were reported in ppm with tetramethylsilane as an internal standard.PLL-mal(y%) with different percentages (y%) of maleimide (from 13% to 26%) were synthesized with a modified procedure of Duan et al.47 (Fig. S1, ESI†). PLL·HBr in PBS buffer (pH 7.0), at a concentration of 10 mg mL−1, was dissolved in 1 mL of PBS (pH 7.4), in order to obtain a final pH of 7.2. The double stoichiometric ratio of NHS-OEG4-maleimide ester was added in sequence to the mixture, under vigorous stirring, and reacted for 4 h at room temperature. Thereafter, the crude mixture was purified using Zeba™ spin desalting columns. For the filtration, the dialyzed solution was removed via centrifugation upon adding 2.5 mL of PBS at pH 7.0 at 1000 rpm for 2 min (twice). Then, repeated twice, 2.5 mL of the desired buffer was added and finally, the polymer solution was filtered. The treated solution was immediately freeze-dried overnight. Afterwards, an NMR spectrum was recorded in D2O with 5 μL of 0.1 M HCl using the water suppression sequence: 1H NMR (400 MHz D2O, pH 6.5) δ [ppm] = 1.26–1.55 ((lysine γ-CH2), 1.63–1.83 (lysine β, δ-H2), 3.00 (free lysine, H2N-CH2), 3.16 (OEG-maleimide, C(O)-NH-CH2–), 4.29 (lysine backbone, NH-CH-C(O)–), 6.86 (maleimide –C(O)-CH-CH-C(O)–) (Fig. S2–S4, ESI†).
Monolayers of PLL-mal(y%) and coupling reaction with CEEEEE
Gold sensors were cleaned by Piranha solution with a ratio of 3:1 (96% H2SO4:30% H2O2) for 1 min, then rinsed with Milli-Q water for 20 min. Thereafter, the surfaces were activated by UV–ozone (UV/Ozone ProCleaner Plus, Bioforce Nanosciences) for 30 min and dipped in PLL-mal(y%) 0.5 mg mL−1 for 30 min. After the washing step with Milli-Q water, PLL-mal(y%) sensors were immersed in CEEEEE 1.0 mM solution for 16 h.
where Ii represents the area of a photoelectron peak and Si the relative sensitivity factor of the peak.
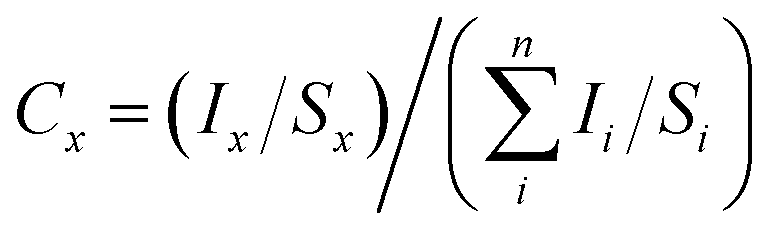
Immobilization of PLL-mal(y%)-CEEEEE monolayer and antifouling measurements by SPRI technique
where Ip and Is refer to the reflected light intensity detected using p- and s-polarized light, respectively. The experiments were carried out by sequentially acquiring 15 frames averaged SPR images with 10 s time delay between them. Kinetic data were obtained by plotting the difference in percent reflectivity (Δ%R) from selected regions of interest (ROIs) of the SPR images as a function of time. The selected ROIs were chosen in order to include all the SPR chip area involved in the surface interaction experiment. All the SPRI experiments were carried out at room temperature.
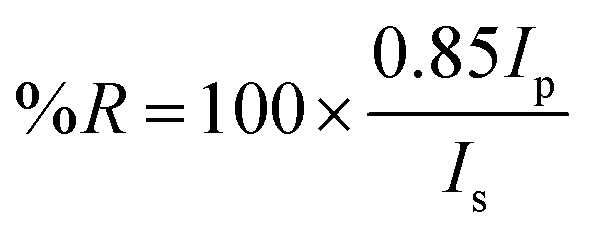
A microfluidic device was used for the analysis: it was fabricated in poly(dimethylsiloxane) (PDMS) polymer through the well-established replica molding technique. The microfluidic device is constituted by six parallel microchannels (200 μm depth, 1.4 cm length, 400 μm width) and circular reservoirs (diameter = 400 μm) at the ends of each channel. PEEK tubes (UpChurch Scientific) were inserted in the circular reservoirs to connect the PDMS microfluidic cell to an Ismatec IPC (Ismatec SA, Switzerland) peristaltic pump. The microfluidic device was built by fixing the PDMS mold on the SPRI gold chip surface. A refractive index matching liquid was employed to obtain the optical contact between the flow cell and the prism.
SPRI gold sensors were rinsed with Milli-Q water, ethanol and dried under N2 for few seconds. Thereafter, the surfaces were activated using UV-ozone for 30 min. The immobilization of the PLL-mal(y%) polymer in PBS buffer 0.01 M, containing 0.150 M NaCl, pH 7.4 was performed on the surface at 20 μL min−1 as flow constant rate for 15 min. PLL-mal(y%) polymer immobilization was followed by the washing step with the same PBS buffer for 10 min. Then, 1.0 mM of oligo CEEEEE in PBS buffer 0.01 M pH 7.5 was immobilized at the same flow rate for 1 hr and 10 min until the visible plateaux of oligo CEEEEE binding on PLL-mal(y%) film.
Immobilization of PLL-mal(y%)-CEEEEE and antifouling measurements by QCM-D technique
:30% H2O2:25% NH4OH 5:1:1) for 5 min, then rinsed with Milli-Q water, dried with a N2 stream and treated with UV-ozone for 10 min. Thereafter, the sensors were placed in the fluid chambers.
Conclusions
A new surface platform based on mixed-charge polymer, peptide–PLL polymers, which enables rapid, direct, effective functionalization of non-fouling layers for biosensing applications was developed in this work. While IR measurements confirmed successful coupling to the maleimide units, peptide–maleimide molar ratios were assessed for the evaluation of the coupling efficiency. All molar ratios were larger than 1:1 for all the y% values of maleimide units, which is in part attributed to differences in hydration between the peptide and the PLL.
Functionalized PLL-mal(26%)-CEEEEE surface revealed significant low-fouling properties in both BSA and human plasma samples through SPRI technique, with 46 ng cm−2 as the value of adsorbed protein when the highest percentage of human plasma samples was utilized. Contrarily, the total nonspecific protein adsorption by QCM-D experiments was greater than 46 ng cm−2 in human plasma, confirming that these measurements are influenced by many factors, such as the hydration shell of bound water molecules co-measured with the plasma proteins, the mechanism of protein adsorption and the molecular size of the plasma proteins. In contrast with many antifouling polymers, the PLL-mal(26%)-CEEEEE films demonstrated an excellent antifouling property using the highest concentration (50.0 mg mL−1) of single-protein solution. In complex media, the PLL-mal(26%)-CEEEEE layer achieved a low-fouling property compared with traditional antifouling coatings. Whether the mechanism of the antifouling behavior works the same as in the zwitterionic materials described above, deserves further investigation. The results obtained for modified-PLL system may have a significant impact on the future development of biosensors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Horizon 2020 Health project “ULTRAPLACAD” (no. 633937) is acknowledged for financial support. A. M. acknowledges financial support from the MESA+Institute for Nanotechnology, University of Twente. N. B. acknowledges support from the Erasmus plus programme for a research stay at the University of Twente.Notes and references
- K. M. Woo, V. J. Chen and P. X. Ma, J. Biomed. Mater. Res., Part A, 2003, 67, 531–537 CrossRef PubMed.
- T. Boontheekul and D. Mooney, J. Curr. Opin. Biotechnol., 2003, 14, 559–565 CrossRef CAS.
- K. Maehashi, T. Katsura, K. Kerman, Y. Takamura, K. Matsumoto and E. Tamiya, Anal. Chem., 2007, 79, 782–787 CrossRef CAS PubMed.
- C. A. Rowe, L. M. Tender, M. J. Feldstein, J. P. Golden, S. B. Scruggs, B. D. MacCraith, J. J. Cras and F. S. Ligler, Anal. Chem., 1999, 71, 3846–3852 CrossRef CAS PubMed.
- X. Liu, L. Yuan, D. Li, Z. Tang, Y. Wang, G. Chen, H. Chen and J. L. Brash, J. Mater. Chem. B, 2014, 2, 5718–5738 RSC.
- Z. Tang, X. Liu, Y. Luan, W. Liu, Z. Wu, D. Li and H. Chen, Polym. Chem., 2013, 4, 5597–5602 RSC.
- P. Vadgama and S. Peteu, Detection Challenges in Clinical Diagnostics, Royal Society of Chemistry, Cambridge, 2013 Search PubMed.
- W. C. Mak, in Biological Fluid-Surface Interactions in Detection and Medical Devices, ed. M. Thompson, C. Blaszykowski, S. C. Rodriguez Emmenegger, A. de los Santos Pereira, Royal Society of Chemistry, Cambridge, 2017, ch. 5, pp. 184–265 Search PubMed.
- E. Luong-Van, I. Rodriguez, H. Y. Low, N. Elmouelhi, B. Lowenhaupt, S. Natarajan, C. T. Lim, R. Prajapati, M. Vyakarnam and K. Cooper, J. Mater. Res., 2013, 28, 165–174 CrossRef CAS.
- E. P. Childers, G. I. Peterson, A. B. Ellenberger, K. Domino, G. V. Seifert and M. L. Becker, Biomacromolecules, 2016, 17, 3396–3403 CrossRef CAS PubMed.
- S. Pasche, J. Vörös, H. J. Griesser, N. D. Spencer and M. Textor, J. Phys. Chem. B, 2005, 109, 17545–17552 CrossRef CAS PubMed.
- S. L. Hirsh, D. R. McKenzie, N. J. Nosworthy, J. A. Denman, O. U. Sezerman and M. M. Bilek, Colloids Surf., B, 2013, 103, 395–404 CrossRef CAS PubMed.
- M. Rabe, D. Verdes and S. Seeger, Adv. Colloid Interface Sci., 2011, 162, 87–106 CrossRef CAS PubMed.
- P. E. Scopelliti, A. Borgonovo, M. Indrieri, L. Giorgetti, G. Bongiorno, R. Carbone, A. Podestà and P. Milani, PLoS One, 2010, 5, e11862 CrossRef PubMed.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- A. Halperin, Langmuir, 1999, 15, 2525–2533 CrossRef CAS.
- P. Vermette and L. Meagher, Colloids Surf., B, 2003, 28, 153–198 CrossRef CAS.
- X. Chen, T. Chen, Z. Lin, X. Li, W. Wu and J. Li, Chem. Commun., 2015, 51, 487–490 RSC.
- X. Chen, M. Yang, B. Liu, Z. Li, H. Tan and J. Li, Langmuir, 2017, 33, 8295–8301 CrossRef CAS PubMed.
- S. Lowe, N. M. O’Brien-Simpson and L. A. Connal, Polym. Chem., 2015, 6, 198–212 RSC.
- S. Chen, L. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef CAS.
- V. B. Damodaran and N. S. Murthy, Biomater. Res., 2016, 20, 18 CrossRef PubMed.
- Q. Shao and S. Jiang, Adv. Mater., 2015, 27, 15–26 CrossRef CAS PubMed.
- C. M. Kirschner and A. B. Brennan, Annu. Rev. Mater. Res., 2012, 42, 211–229 CrossRef CAS.
- C. Leng, H. Huang, K. Zhang, H. C. Hung, Y. Xu, Y. Li, S. Jiang and Z. Chen, Langmuir, 2018, 34, 6538–6545 CrossRef CAS PubMed.
- F. Wan, Q. Ye and F. Zhou, in Antifouling of Micro-/Nanostructural Surfaces, ed. F. Zhou, Springer, Berlin, Heidelberg, 2015, ch. 4, pp. 83–104 Search PubMed.
- C. M. Kirschner and A. B. Brennan, Annu. Rev. Mater. Res., 2012, 42, 211–229 CrossRef CAS.
- S. Chen, F. Yu, Q. Yu, Y. He and S. Jiang, Langmuir, 2006, 22, 8186–8191 CrossRef CAS PubMed.
- H. Vaisocherová, W. Yang, Z. Zhang, Z. Cao, G. Cheng, M. Piliarik, J. Homola and S. Jiang, Anal. Chem., 2008, 80, 7894–7901 CrossRef PubMed.
- J. Ladd, Z. Zhang, S. Chen, J. C. Hower and S. Jiang, Biomacromolecules, 2008, 9, 1357–1361 CrossRef CAS PubMed.
- Z. Zhang, S. Chen and S. Jiang, Biomacromolecules, 2006, 7, 3311–3315 CrossRef CAS PubMed.
- G. Cheng, H. Xue, Z. Zhang, S. Chen and S. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8831–8834 CrossRef CAS PubMed.
- R. E. Holmlin, X. X. Chen, R. G. Chapman, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 2841–2850 CrossRef CAS.
- M. Bernards, G. Cheng, Z. Zhang, S. Chen and S. Jiang, Macromolecules, 2008, 41, 4216–4219 CrossRef CAS.
- S. Chen and S. Y. Jiang, Adv. Mater., 2008, 20, 335–338 CrossRef CAS.
- J. K. West, R. Latour and L. L. Hench, J. Biomed. Mater. Res., 1997, 37, 585–591 CrossRef CAS PubMed.
- M. Schuler, G. R. Owen, D. W. Hamilton, M. de Wild, M. Textor, D. M. Brunette and S. G. P. Tosatti, Biomaterials, 2006, 27, 4003–4015 CrossRef CAS PubMed.
- S. Lee and N. D. Spencer, Langmuir, 2008, 24, 9479–9488 CrossRef CAS PubMed.
- S. G. Zhu, J. J. Xiang, X. L. Li, S. R. Shen, H. Lu, J. Zhou, W. Xiong, B. C. Zhang, X. M. Nie and M. Zhou, et al. , Biotechnol. Appl. Biochem., 2004, 39(2), 179–187 CrossRef CAS PubMed.
- D. Mazia, G. Schatten and W. Sale, J. Cell Biol., 1975, 66, 198–200 CrossRef CAS PubMed.
- S. Gac-Breton, J. Coudane, M. Boustta and M. Vert, J. Drug Targeting, 2004, 12, 297–307 CrossRef CAS PubMed.
- Y. H. Choi, F. Liu, J. S. Kim, Y. K. Choi, P. Jong Sang and S. W. Kim, J. Controlled Release, 1998, 54, 39–48 CrossRef PubMed.
- H. K. Sun, H. J. Ji, O. J. Cheol and G. P. Tae, J. Controlled Release, 2005, 103, 625–634 CrossRef PubMed.
- S. R. Hynes, M. F. Rauch, J. P. Bertram and E. B. Lavik, J. Biomed. Mater. Res., Part A, 2009, 89, 499–509 CrossRef PubMed.
- S. Morgenthaler, C. Zink, B. Städler, J. Vörös, S. Lee, N. D. Spencer and S. G. Tosatti, Biointerphases, 2006, 1, 156 CrossRef CAS PubMed.
- Y. Luan, D. Li, T. Wei, M. Wang, Z. Tang, J. L. Brash and H. Chen, Anal. Chem., 2017, 89, 4184–4191 CrossRef CAS PubMed.
- X. Duan, L. Mu, S. D. Sawtelle, N. K. Rajan, Z. Han, Y. Wang, H. Qu and M. A. Reed, Adv. Funct. Mater., 2015, 25, 2279–2286 CrossRef CAS.
- J. Kyte and R. F. Doolittle, J. Mol. Biol., 1982, 157, 105–132 CrossRef CAS PubMed.
- B. L. Frey and R. M. Corn, Anal. Chem., 1996, 68, 3187–3193 CrossRef CAS.
- C. E. Jordan, A. G. Frutos, A. J. Thiel and R. M. Corn, Anal. Chem., 1997, 69, 4939–4947 CrossRef CAS.
- S. Sheikh, D. Y. Yang, C. Blaszykowski and M. Thompson, Chem. Commun., 2012, 48, 1305–1307 RSC.
- S. W. Tam-Chang, H. A. Biebuyck, G. M. Whitesides, N. Jeon and R. G. Nuzzo, Langmuir, 1995, 11, 4371–4382 CrossRef CAS.
- T. M. Blattler, S. Pasche, M. Textor and H. J. Griesser, Langmuir, 2006, 22, 5760–5769 CrossRef PubMed.
- M. Volmer-Uebing and M. Stratmann, Appl. Surf. Sci., 1992, 55, 19–35 CrossRef CAS.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083–5086 CrossRef CAS.
- I. Reviakine, D. Johannsmann and R. P. Richter, Anal. Chem., 2011, 83, 8838–8848 CrossRef CAS PubMed.
- H. Wiig, O. Kolmannskog, O. Tenstad and J. L. Bert, J. Physiol., 2003, 550, 505–514 CrossRef CAS PubMed.
- P. Akkahat, S. Kiatkamjornwong, S. Yusa, V. P. Hoven and Y. Iwasaki, Langmuir, 2012, 28, 5872–5881 CrossRef CAS PubMed.
- C. Situ, A. R. Wylie, A. Douglas and C. T. Elliott, Talanta, 2008, 76, 832–836 CrossRef CAS PubMed.
- K. Uchida, Y. Hoshino, A. Tamura, K. Yoshimoto, S. Kojima, K. Yamashita, I. Yamanaka, H. Otsuka, K. Kataoka and Y. Nagasaki, Biointerphases, 2008, 2, 126–130 CrossRef PubMed.
- H. Vaisocherová, E. Brynda and J. Homola, Anal. Bioanal. Chem., 2015, 407, 3927–3953 CrossRef PubMed.
- N. Xia, L. Liu, M. G. Harrington, J. Wang and F. Zhou, Anal. Chem., 2010, 82, 10151–10157 CrossRef CAS PubMed.
- F. Fernandez, K. Hegnerova, M. Piliarik, F. Sanchez-Baeza, J. Homola and M. P. Marco, Biosens. Bioelectron., 2010, 26, 1231–1238 CrossRef CAS PubMed.
- H. Vaisocherová, V. Sevcu, P. Adam, B. Spackova, K. Hegnerova, A. de los Santos Pereira, C. Rodriguez-Emmenegger, T. Riedel, M. Houska, E. Brynda and J. Homola, Biosens. Bioelectron., 2014, 51, 150–157 CrossRef PubMed.
- T. Riedel, Z. Riedelova-Reicheltova, P. Majek, C. Rodriguez-Emmenegger, M. Houska, J. E. Dyr and E. Brynda, Langmuir, 2013, 29, 3388–3397 CrossRef CAS PubMed.
- J. S. Shumaker-Parry and C. T. Campbell, Anal. Chem., 2004, 76, 907–917 CrossRef CAS PubMed.
- L. S. Jung, C. T. Campbell, T. M. Chinowsky, M. N. Mar and S. S. Yee, Langmuir, 1998, 14, 5636–5648 CrossRef CAS.
- H. Fischer, I. Polikarpov and A. F. Craievich, Protein Sci., 2004, 13, 2825–2828 CrossRef CAS PubMed.
- J. Vörös, Biophys. J., 2004, 87, 553–561 CrossRef PubMed.
- H. Zhao, P. H. Brown and P. Schuck, Biophys. J., 2011, 100, 2309–2317 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR and PM-IRRAS data. See DOI: 10.1039/c8tb01619d |
| This journal is © The Royal Society of Chemistry 2018 |