
Enhancing the sensitivity of colorimetric lateral flow assay (CLFA) through signal amplification techniques
 a
and
Xiaohu
Xia
*a
a
and
Xiaohu
Xia
*a
Abstract
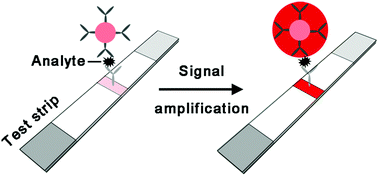
Colorimetric lateral flow assay (CLFA) is one of a handful of diagnostic technologies that can be truly taken out of the laboratory for point-of-care testing without the need for any equipment and skilled personnel. Despite its simplicity and practicality, it remains a grand challenge to substantially enhance the detection sensitivity of CLFA without adding complexity. Such a limitation in sensitivity inhibits many critical applications such as early detection of significant cancers and severe infectious diseases. With the rapid development of materials science and nanotechnology, signal amplification techniques that hold great potential to break through the existing detection limit barrier of CLFA have been developed in recent years. This article specifically highlights these emerging techniques for CLFA development. The rationale behind and advantages and limitations of each technique are discussed. Perspectives on future research directions in this niche and important field are provided.
- This article is part of the themed collections: Recent Review Articles and Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

