
Binding of an amphiphilic phthalocyanine to pre-formed liposomes confers light-triggered cargo release†

Abstract
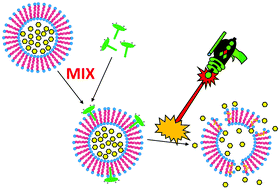
Liposomes are able to load a range of cargos and have been used for drug delivery applications, including for stimuli-triggered drug release. Here, we describe an approach for imparting near infrared (NIR) light-triggered release to pre-formed liposomes, using a newly-synthesized cationic, amphiphilic phthalocyanine. When simply mixed in aqueous solution with cargo-loaded liposomes, the cationic amphiphilic phthalocyanine, but not a cationic hydrophilic azaphthalocyanine, spontaneously incorporates into the liposome bilayer. This enables the subsequent release of loaded cargo (doxorubicin or Basic Orange) upon irradiation with NIR light. The rate of release could be altered by varying the amount of photosensitizer added to the liposomes. In the absence of NIR light exposure, stable cargo loading of the liposomes was maintained.
- This article is part of the themed collection: Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

