
Enhancing anti-thrombogenicity of biodegradable polyurethanes through drug molecule incorporation†
 *ab
*ab
Abstract
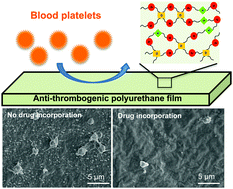
Sufficient and sustained anti-thrombogenicity is essential for blood-contacting materials because blood coagulation and thrombosis caused by platelet adhesion and activation on material surfaces may lead to functional failure and even fatal outcomes. Covalently conjugating anti-thrombogenic moieties into a polymer, instead of surface modification or blending, can maintain the anti-thrombogenicity of the polymer at a high level over time. In this study, a series of randomly crosslinked, elastic, biodegradable polyurethanes (PU-DPA) were synthesized through a one-pot and one-step method from polycaprolactone (PCL) diol, hexamethylene diisocyanate (HDI) and an anti-thrombogenic drug, dipyridamole (DPA). The mechanical properties, hydrophilicity, in vitro degradation, and anti-thrombogenicity of the resultant PU-DPA polymers could be tuned by altering the incorporated DPA amount. The surface and bulk hydrophilicity of the polyurethanes decreased with increasing hydrophobic DPA amounts. All PU-DPA polymers exhibited strong mechanical properties and good elasticity. The degradation rates of PU-DPAs decreased with increasing DPA content in both PBS and lipase/PBS solutions. Covalently incorporating DPA into the polyurethane significantly reduced the platelet deposition compared to that of the polyurethane without DPA, and the polyurethane remained anti-thrombogenicity after degradation. The PU-DPA films also supported the growth of human umbilical vein endothelial cells. The attractive mechanical properties, blood compatibility, and cell compatibility of this anti-thrombogenic biodegradable polyurethane indicate that it has a great potential to be utilized for blood-contacting devices and cardiovascular tissue repair and regeneration.
- This article is part of the themed collection: Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

