
Pharmaceutical crystallization in surface-modified nanocellulose organogels†

Abstract
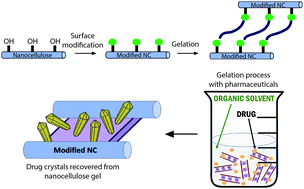
A significant research focus in the pharmaceutical industry is on methods to improve drug uptake into the body by increased dissolution of poorly water soluble active pharmaceutical ingredients (APIs) or sustained drug release behavior, which results in higher overall uptake. Production of higher energy, higher solubility polymorphs is one approach to address this problem. Here we utilize natural materials, cellulose nanocrystals (CNCs), that have a high surface area covered with readily-modified hydroxyl groups to form organogels that promote API crystallization into polymorphs that differ from the as-received materials. We form the gels by oxidizing the CNCs and mixing them with an amine-containing surfactant, octadecylamine (ODA) in dimethylsulfoxide (DMSO) and we optimize the composition and preparation conditions for these gels. The APIs, sulfamethoxazole, sulfapyridine, and sulfamerazine, are added to the mixture prior to the gelation step and are expected to localize in the solvophobic regions of the physical gel and crystallize. We found that sulfamethoxazle recovered from the gels is in the amorphous state, while sulfapyridine crystallizes into a mixture of forms I, III and IV, and sulfamerazine crystallizes into forms I and II, which are different from the as-received materials. This system shows promise for rational design of nanocellulose organogel supports for heterogeneous crystallization of pharmaceutical materials with desired polymorphs.
- This article is part of the themed collections: Materials and Nano Research in Atlanta and Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

