
Statistical versus block fluoropolymers in gene delivery†
 *a
*a
Abstract
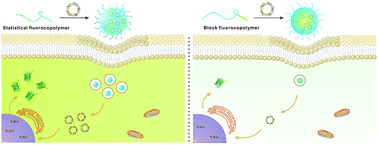
Fluoropolymers have shown great promise in non-viral gene delivery. The current fluoropolymers developed for gene delivery are synthesized by grafting fluoroalkyls or fluoroaromatics onto cationic polymers. To expand the family of fluoropolymers for the transduction of nucleic acids, new strategies to synthesize fluoropolymers are required. In this study, we synthesized both statistical and block copolymers of poly(2-dimethylaminoethyl methacrylate) (pDMAEMA) and poly(heptafluorobutyl methacrylate) (pHFMA) via reversible addition–fragmentation chain transfer polymerization, and the transfection efficiencies of the fluorocopolymers were evaluated. The statistical fluorocopolymer exhibited dramatically higher performance in gene delivery than the block one, which is attributed to more efficient and sustained DNA uptake by the transfected cells. Moreover, the statistical copolymer of DMAEMA and HFMA showed a fluorine effect in gene delivery, and its efficiency was much superior to non-fluorinated polymers. The results revealed the structure and activity relationships of fluoropolymers consisting of DMAEMA and HFMA, and provided a new insight to guide the design of fluoropolymers for efficient gene delivery.
- This article is part of the themed collection: Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

