
Biomolecular dynamic covalent polymers for DNA complexation and siRNA delivery†

Abstract
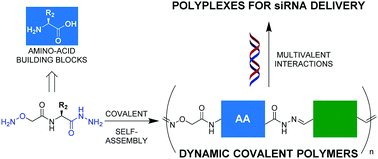
Synthetic delivery systems that are described as smart are considered essential for the successful development of gene therapies. Dynamic covalent polymers (DCP) are dynamic and adaptive species that can expand and shorten their main chain in a reversible fashion. In particular, polyacylhydrazone DCPs are pH-sensitive and undergo hydrolytic dissociation at acidic pH, which is an interesting feature for gene delivery. Building upon our previous finding that cationic DCPs can complex DNA through multivalent interactions, we report here on a new generation of DCPs that incorporate modified amino acids. The covalent self-assembly through polycondensation was extended towards multifunctional DCPs combining different building blocks and different molecular dynamics. These biomolecular DCPs were found able to complex both long DNA and siRNA, and biological studies demonstrate that they are able to deliver functional siRNA in living cells. This straightforward and modular approach to the self-production of multifunctional and biomolecular DCPs as siRNA vectors can therefore constitute a stepping stone in smart gene delivery using dynamic and adaptive biodynamers.
- This article is part of the themed collection: Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

