
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid formulation of redox-responsive oligo-β-aminoester polyplexes with siRNA via jet printing†
Tatiana
Lovato‡
a,
Vincenzo
Taresco‡
a,
Ali
Alazzo
a,
Caterina
Sansone
b,
Snjezana
Stolnik
 a,
Cameron
Alexander
*a and
Claudia
Conte
*ab
a,
Cameron
Alexander
*a and
Claudia
Conte
*ab
aDivision of Molecular Therapeutics and Formulation, School of Pharmacy, University of Nottingham, NG7 2RD, UK. E-mail: cameron.alexander@nottingham.ac.uk
bDrug Delivery Laboratory, Department of Pharmacy, University of Napoli Federico II, Via Domenico Montesano 49, 80131 Napoli, Italy. E-mail: claudia.conte@unina.it
First published on 9th October 2018
Abstract
Here we describe a rapid inkjet formulation method for screening newly-synthesised cationic materials for siRNA delivery into cancer cells. Reduction responsive oligo-β-aminoesters were prepared and evaluated for their ability to condense siRNA into polyplexes through a fast inkjet printing method. A direct relationship between the oligomer structures and charge densities, and the final cell response in terms of uptake rate and transfection efficacy, was found. The oligo-β-aminoesters were well-tolerated by the cancer cells, compared to conventional cationic polymers so far employed in gene delivery, and were as active in silencing of a representative luciferase gene.
Introduction
Successful and practical gene therapies require consistent formulations which can be easily prepared, and which can effectively deliver nucleic acids to intracellular target sites.1,2 In turn these formulations need nucleic acid carrier vehicles which are safe in human patients, and with the highest performance in terms of in vivo stability, cellular delivery and transfection efficiency.3 Accordingly, combinations of advances in materials chemistry and pharmaceutical formulations are pre-requisites for nucleic acid therapeutics.4To date, the formulation parameters for nucleic acid delivery have generally evolved from research lab-based protocols for preparing polymer–nucleic acid polyelectrolyte complexes, and these can be highly ‘operator-dependent’. In part this is due to the many variables important in the polycation carrier/nucleic acid polyanion association process, as the kinetics of polyelectrolyte complex formation are highly variable across concentration ranges, order and speed of addition.5–8 As a consequence, methods to prepare, formulate and screen cationic polymers for nucleic acid delivery are required, and these protocols need to be rapid, use small quantities and be easily applicable across ranges of physical and chemical properties. The increasing use of printing methods for screening pharmaceutical materials is a potential means by which polymer–nucleic acid complexes might be optimised for practical formulations.9–11
Concurrently, a very wide range of natural and synthetic cationic polymers have been explored as potential non-viral gene delivery systems, particularly for cancer treatment.1 Amongst these materials, oligo- and poly(β-aminoester)s (OBAEs and PBAEs) have emerged as promising candidates, as they are easy to synthesise and are biodegradable due to their hydrolysable ester backbones.12,13 These materials are generally positively charged at physiological pH and thus are able to condense spontaneously with nucleic acids through electrostatic interactions. The main advantage of PBAEs compared to other conventional cationic materials is their significantly lower cytotoxicity, combined with their ability to transfect cells with high efficiency.14,15 However, the molecular weight, the structure and the supramolecular architectures of polycations, including PBAEs, play a pivotal role in the final biological effect, influencing the cytotoxicity as well as the gene transfection activity.16–20 For instance, it has been demonstrated that polycations with high molecular weight (MW) usually show appreciable cytotoxicities, even though their stronger condensation capacity toward nucleic acids tends to improve transfection potency compared to their lower molar mass analogues.21,22 In addition, dependent on their ratio of primary, secondary and tertiary amines, OBAEs and PBAEs may display appropriate pKa ranges to exploit the “proton sponge mechanism” thus enhancing escape of polyplexes and nucleic acids from endosomal compartments which in turn improves access to the targeted genes.18,23,24 However, while highly stable polymer–DNA complexes are desirable during the initial stages of the delivery process, the release of the nucleic acid cargo is more rapid if the vector can be degraded into smaller, less charged, fragments. The hydrolysis of the polyester backbones in PBAEs in cellular fluids typically occurs on the time scale of several hours to a few days, depending on the polymer structure, thus affecting the release of the gene cargoes and the final transfection effect. While it is possible to tune the degradation rate of the PBAE polymer via molar mass and monomer structure, it is also desirable to encode “on-demand” cargo release, such that very rapid dissociation of the nucleic acid can occur at the correct cellular region. This can be achieved by incorporating disulfide bonds in the polymer backbone, which can be cleaved in the reducing environments of certain intracellular milieu, thereby improved the efficiency of gene delivery.25,26
In this manuscript, we report the synthesis of a small range of redox responsive, cytocompatible oligo-β-aminoesters (OBAEs) which are able to condense and transfect siRNA into cancer cells. Through a specific modulation of the molar ratio between the starting materials, we obtained OBAEs with different structures and positive charge densities, thus tuning their capabilities to interact with siRNA and elicit a subsequent biological response. Furthermore, we designed the OBAEs to have solubility properties allowing them to be formed into-siRNA polyplexes via an easy, fast and cheap inkjet technology. We considered inkjet printing to be particularly suitable for screening the formulations of biotherapeutics in this study as this technique is versatile, scalable and can deposit picolitre volumes of solution with high accuracy and reproducibility. The data together show that oligomeric cations and siRNA can be easily print-formulated into effective in vitro nucleic acid delivery systems.
Experimental
Materials
All solvents were of analytical or HPLC grade and purchased from Sigma or Fisher Scientific unless otherwise stated. Deuterated solvents were from Sigma. Acryloyl chloride, ethylene-dioxy-bis-ethylamine, triethylamine (TEA), dithiodiethanol, sodium chloride, potassium phosphate dibasic and potassium phosphate monobasic, sodium azide, sodium phosphate dibasic, ethidium bromide (EtBr), fluorescamine, polyethylenimine (PEI) (25 kDa, branched) calf thymus DNA and glutathione (GSH), were used as received from Sigma Aldrich. Luciferase siRNA (CCGCAAGAUCCGCGAGAUU) was provided by Eurogentec (UK). Luciferase Assay System with Reporter Lysis Buffer and CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) were provided by Promega (UK). Silencer™ Cy™3-labeled Negative Control was provided by Thermofisher Scientific (UK).Synthesis and characterization of cationic oligo (β-amino ester)s
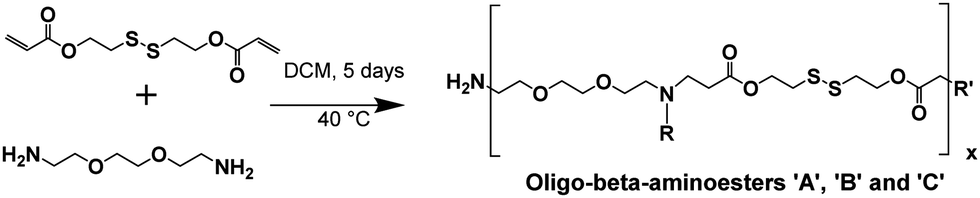
The synthesis of the oligo-β-aminoester (OBAE) was via a two-step reaction, i.e. (1) the synthesis of disulfanediylbis(ethane-2,1-diyl) diacrylate and (2) the Michael addition reaction between disulfanediylbis(ethane-2,1-diyl) diacrylate and ethylenedioxy-bis-ethylamine.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 as eluents to generate a soft solid (yield 85%).
:10 as eluents to generate a soft solid (yield 85%).
ATR-IR: ν (cm−1) 3449, 2978, 2874, 1725, 1634, 1620, 1513, 1454, 1408, 1371, 1223, 1177, 1045, 978, 911, 814, 663.
1H NMR (400 MHz, d6-DMSO, ppm): δH 3.01–3.03 (t, 2, J = 8 Hz, –SSCH2CH2O–), 4.35–4.38 (t, 2, J = 4 Hz, –SSCH2CH2O–), 5.95 (dd, 1, J = 4 Hz, J = 8 Hz, –OCOCHCH2), 6.17 (dd, 1, J = 8 Hz, J = 16 Hz, –OCOCHCH2), 6.36 (dd, 1, J = 4 Hz, J = 20 Hz, –OCOCHCH2).
13C NMR (400 MHz, d6-DMSO, ppm): δC 36.4 (s, 1, –SSCH2CH2O–), 62.1 (s, 1, –SSCH2CH2O–), 128.1 (s, 1, –OCOCHCH2), 131.8 (s, 1, –OCOCHCH2), 165.0 (s, 1, –OCOCHCH2).
ATR-IR: ν (cm−1) 3269, 3070, 2865, 1643, 1551, 1494, 1457, 1355, 1295, 1099, 1024, 819, 755.
1H NMR (500 MHz, d6-DMSO, ppm): δH 2.50–2.53 (m, 2, –OCOCH2CH2–), 2.72–2.79 (m, 2, –NCH2CH2O–), 2.78–2.81 (t, 2, J = 15 Hz, –SSCH2CH2O–), 2.94–3.04 (m, 2, –OCOCH2CH2–), 3.21–3.24 (m, 2, –OCH2CH2NH2), 3.41–3.44 (m, 2, –OCH2CH2NH2), 3.54–3.57 (m, 2, –NCH2CH2O–), 3.58–3.60 (m, 4, –OCH2CH2O–, –OCH2CH2O–), 3.61–3.63 (m, 2, –SSCH2CH2O–), 8.27 (br, 3, –OCH2CH2NH3+).
13C NMR (500 MHz, d6-DMSO, ppm): δC 32.4 (s, 1, –OCOCH2CH2–), 39.0 (s, 1, –OCH2CH2NH2–), 41.5 (s, 2, –SSCH2CH2O–), 44.3 (s, 1, –OCOCH2CH2–), 51.4 (s, 1, –NCH2CH2O–), 60.1 (s, 2, –SSCH2CH2O–), 69.4 (s, 2, –OCH2CH2O–, –OCH2CH2O–), 69.8 (s, 1, –OCH2CH2NH2–), 70.0 (s, 1, –NCH2CH2O–), 170.3 (s, 2, –OCOCH2CH2–) m/z found [M − H]− (A): 2331; (B): 1434; (C): 560.
Characterization of OBAE
1H-, 13C-NMR, 2D-NMR (COSY, HSQC, HMBC) spectra were recorded at 25 °C on a Bruker Advance III 500 MHz spectrometer. All chemical shifts are reported in ppm (δ) relative to tetramethylsilane or referenced to the chemical shifts of residual solvent resonances. Multiplicities are described with the following abbreviations: s = singlet, br = broad, d = doublet, t = triplet, m = multiplet, dd = doublet of doublets. MestReNova 6.0.2 copyright 2009 (Mestrelab Research S. L.) was used for analysing the spectra.FT-IR spectra were recorded with an attenuated total reflection spectrophotometer (Agilent Technologies Cary 630 FTIR) equipped with a diamond single reflection ATR unit. Spectra were acquired with a resolution of 4 cm−1 by co-adding 32 interferograms, in the range 4000–650 cm−1.
IR analysis were performed by using SpectraGryph version1.0.
Mass spectra were carried out using a Micromass LCT ToF with electrospray ionization and OpenLynx software.
Fluorescamine assays for amine content determination
The intensity of fluorophore resulting from the reaction of fluorescamine with primary amine was measured using a Cary Eclipse Fluorescence Spectrophotometer and glycine as standard. To a 2 mL sample of polymer solution (concentration 10 μg mL−1, in borate buffer pH 8.7), 0.5 mL of fluorescamine in acetone (0.3 mg mL−1) were added and vortexed for 10 seconds. After incubation of the reagents in the dark for 20 minutes, the emission from the resulting solution was measured at a wavelength of 480 nm using an excitation wavelength of 385 nm against a blank of borate buffer with fluorescamine.Buffer capacity of OBAE
The buffer capacity of OBAE was determined by acid–base titration. Acid–base titration was performed using a Fisherbrand pH meter with a Hydrus 600 electrode. Briefly, 0.5 mg of PBAE were dissolved in 1 mL of NaCl 0.1 M and the pH of the polymer solution was adjusted with 0.1 M HCl to 3. Then, the solution was titrated with NaOH 0.1 M and the pH value of solution was measured.The buffering capacity was defined as the percentage of the protonated amine groups from pH 7.4 to 5.0 and calculated according to the following equation:
| Buffer capacity (%) = 100(ΔVNaOH × 0.1 M)/N mol |
For total amine content, the volume of NaOH required to ionize all the amine groups based on the first derivative analysis of titration curve was used and multiplied by the concentration (0.1 M). As the concentration of NaOH is the same, it can be removed from the above equation.
Printing set-up and work flow conditions
Prior to dispensing the siRNA aqueous solutions into a 96-wellplate filled with 100 μL of water solutions of OBAEs for each well, the target had to be defined. Firstly, the outer dimensions of the wellplate were added to the software sciFLEXARRAYER (Scienion AG, version 2.09.002) followed by defining the number of wells, well distance, well depth and the spot area (area within the target designated for spotting). SiRNA solutions were dispensed via a piezo electric inkjet printer (Sciflexarray S5, Scienion) through a 90 μm orifice nozzle. Each droplet was dispensed with a rate of 30–40 drop per μs with sizes ranging in between 250 and 280 pL. Droplet volume was altered by tuning the values of the voltage (98–105 volt) and electrical pulse (45–55 μs). Images of the drop formation and droplet size were obtained using the printer software. The final spots were imaged using the Leica MZ16 stereomicroscope.Depending on the siRNA/oligomer ratios to be reached, the number of drops per spot was adjusted accordingly. The nozzle was washed with Milli-Q water, in between each printing cycle, as part of the automated printing-washing loop. The nozzle was programmed to dispense the siRNA solutions into the well from a vertical distance of circa 10–20 mm from the well-plate, no contact between the nozzle tip and the water surface was allowed.
Polyplexes characterization
The hydrodynamic diameter (DH), polydispersity index (PI) and zeta potential of polyplexes were determined on a Zetasizer Nano ZS (Malvern Instruments Ltd). A NP dispersion was diluted in Milli-Q water at intensity in the range 104–106 counts per s and measurements were performed at 25 °C on 90° angle. Results are reported as mean of three separate measurements of three different batches (n = 9) ± standard deviation (SD). Morphology and shape of polyplexes were monitored through Atomic Force Microscopy (Bruken Dimension Fast Scan Bio).SiRNA complexation was confirmed by agarose gel retardation. Polyplexes containing 1 μg of siRNA were loaded on 2% agarose gel in Tris–Acetate–EDTA (TAE) buffer and subjected to electrophoresis for 45 min at 70 V. SiRNA bands were stained with EtBr and finally visualized with an UV illuminator.
Ethidium bromide displacement assay
In this assay, calf thymus DNA (50 μg mL−1 in PBS) was incubated with ethidium bromide (2 μg mL−1) for 30 minutes. Thereafter, aliquots of 100 μL were mixed with 100 μL of polymer solutions at different concentrations. The samples were incubated under stirring for 30 minutes, and then the fluorescence intensity of DNA–ethidium bromide complexes was measured at a wavelength of 590 nm using an excitation wavelength of 520 nm.Cell culture
A549 lung cancer cells were obtained from the American Type Culture Collection, cultured at 37 °C in a humidified atmosphere containing 5% CO2 and grown continuously in DMEM supplemented with 10% FBS, 100 unit per mL penicillin and 100 μg mL−1 streptomycin.Cell metabolic activity (viability) assay
A549 luciferase expressing cells (2 × 104) were placed in 96-well plates and cultured in 200 μL of cell medium with or without FBS at 10%. After 24 h, cells were treated with free OBAEs in the concentration range 0.005–5 mg mL−1. As control, cells were treated as well as with an aqueous solution of free polyethylenimine (PEI) at the same concentration range. Cells treated with 0.1% (v/v) Triton-X 100 and fresh media were used as a positive and a negative control, respectively. After 24 h of incubation, cells were washed with PBS and treated with CellTiter 96® Aqueous One Solution Cell Proliferation Assay (MTS, Promega) (20 μL per well). After further incubation (3 h), the absorbance was read at 490 nm in a microplate reader (Tecan Platereader). The percentage of metabolic activity (%) was calculated according to the equation:| Cell viability (%) = [(OD sample − OD CTR+)/(OD CTR − OD CTR+)] × 100 |
Transfection studies
For transfection studies, A549 luciferase expressing cells (5 × 104) were seeded into 24-well plates and cultured in 500 μL of cell medium with FBS at 10%. After 24 h, the culture medium was replaced with 0.5 mL of fresh serum-free DMEM and treated with siRNA/OBAE polyplexes containing 1 μg of siRNA/well. After 4 h of incubation, cells were washed three times with fresh medium in order to remove NPs and incubated again at 37 °C until 48 h. Finally, after transfection, luciferase activity was measured as RLU per mg protein using the luciferase assay system (Luciferase Assay System with Reporter Lysis Buffer, Promega) and BCA reagent (Sigma, UK).Uptake and endolysosomal escape
A549 luciferase expressing cells (2 × 104) were seeded in eight well chamber slides (Nunc, Thermo Fisher Scientific Inc., UK) and cultured in 300 μL of cell medium with FBS at 10%. After 24 h, the culture medium was replaced with 0.3 mL of fresh serum-free DMEM and treated with 10 mg mL−1 of polyplexes formed by OBAEs and Cy3-siRNA at 10:1 polymer/siRNA weight ratio. After 4 h of incubation, cells were washed three times with fresh medium without Phenol Red and treated with Lysotracker Green DND-26 (Invitrogen Life Technologies, UK) containing media (100 nM) and then with DAPI (Invitrogen Life Technologies, UK) containing media (1 μg mL−1) for 10 minutes at rt in the dark according to the manufacturer's specifications. Live cells were finally imaged through confocal microscopy (Zeiss LSM 700 Confocal Laser Scanning Microscope equipped with argon and HeNe lasers and a 40×/1.2 NA water objective). Zen 2009 image Software was utilized for image processing.
Statistical analysis
Unless otherwise stated, all data are shown as mean ± standard deviation (SD), two-way analysis of variance (ANOVA) was applied for comparison of three or more group means (Tukey's multiple comparisons test). P value of <0.05 was considered statistically significant. ****, ***, **, and * display p < 0.0001, p < 0.001, p < 0.01, and p < 0.05, respectively. GraphPad Prism 6 software was used for data analysis.Results and discussion
The initial focus for the study was to develop oligomers and polymers with properties suitable for rapid condensation of siRNA via inkjet printing and also triggered degradation properties. Accordingly, OBAEs were synthesized through Michael-type addition reaction between the acrylate groups of a disulfanediylbis(ethane-2,1-diyl)diacrylate (DSD), and the terminal amino groups of ethylene-dioxy-bis-ethylamine.The bioreducible disulfide-containing monomer DSD was synthesized through reaction of dithiodiethanol with acryloyl chloride and its structure confirmed by 1H, 13C and 2D-NMR spectroscopy (Fig. S1, ESI†), as previously reported.27 Then, DSD was employed in a aza-Michael addition reaction with the diamine ethylen-bioxy-bis-ethylamine, using different molar ratios of the starting materials (1:1, 1:1.25 and 1:1.5 DSD/ethylene-dioxy-bis-ethylamine ratio), thus yielding three different OBAEs (A, B and C, respectively). The ratios of the diacrylate and diamine were varied in order to modulate the final properties of the products. The higher diamine ratio was intended to produce a lower molar mass product and the 1:1 mixture the highest molar mass, which we anticipated would affect their ‘printability’.
The condensation reactions were allowed to proceed for 5 days at 30 °C (Fig. 1). All the OBAEs synthesized were characterized through 1H, 13C and 2D-NMR spectroscopy in d6-DMSO (Fig. 2 and Fig. S2, S3, ESI†), Electron Spray Ionization (ESI) mass spectra (Fig. S5, ESI†) and FT-ATR-IR spectroscopy (Fig. S6, ESI†).
 | ||
| Fig. 1 Synthetic reaction of OBAEs. | ||
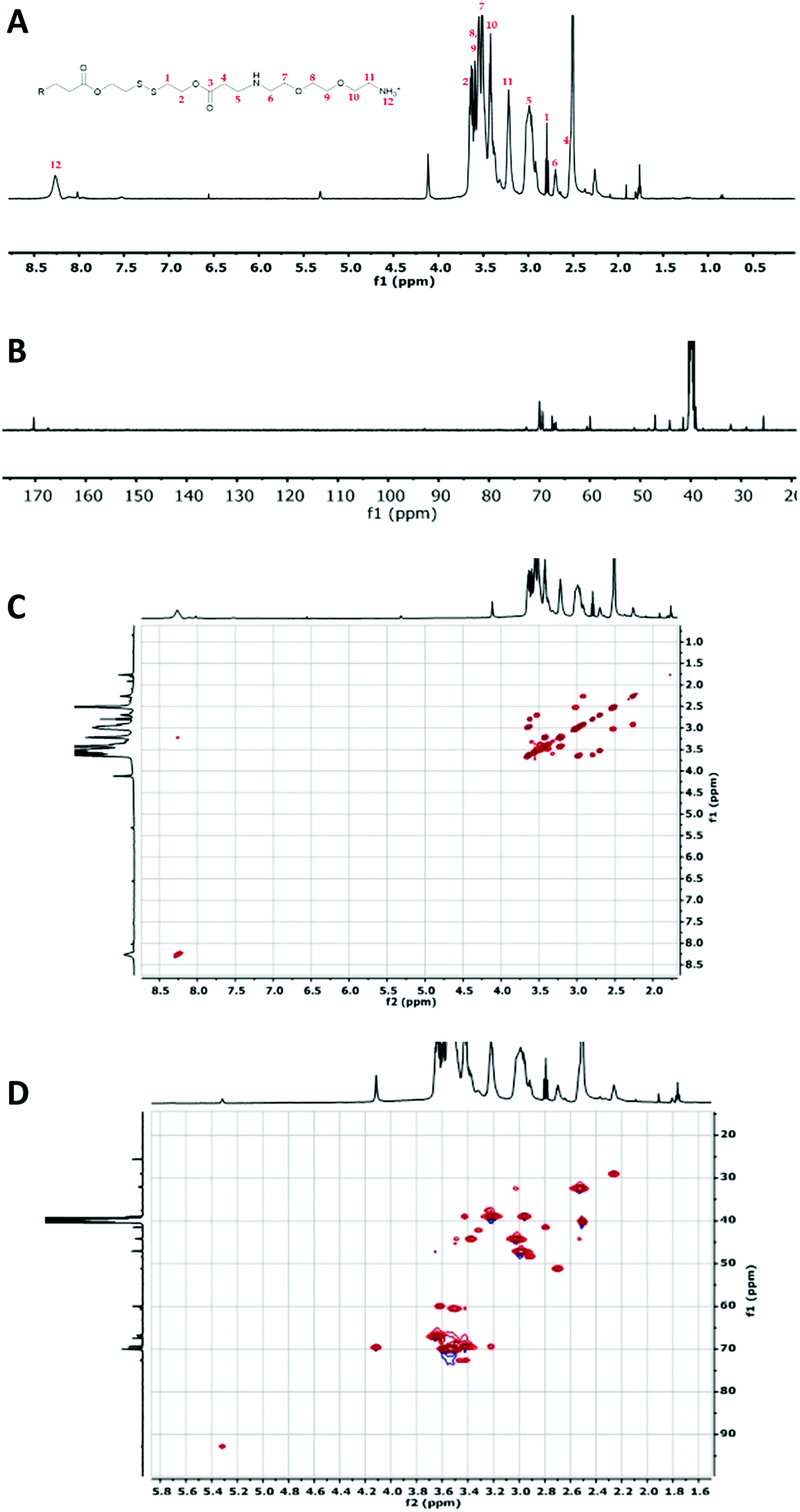 | ||
| Fig. 2 Characterization of OBAE C. (A) 1H NMR spectrum; (B) 13C NMR spectrum collected for 6 h; (C) 1H–1H COSY NMR spectrum and (D) 1H–13C HSQC NMR spectrum. Spectra recorded at 500 MHz in d6-DMSO. | ||
The 1H-NMR spectra of the three OBAEs synthesised gave very similar NMR spectra (Fig. 2 and Fig. S2A, S3A, ESI†). In particular, the resonances of the protons of the methyl groups at δ = 2.25–2.27 ppm and δ = 2.90–3.02 ppm (no. 5 and 6 respectively) confirmed the presence of a secondary amine in the OBAE backbone. The signal of the protons of the methyl groups adjacent to the disulfide bond at δ = 3.41–3.43 ppm (no. 1) demonstrated the integrity of the disulfide bond after the reaction with ethylene-dioxy-bis-ethylamine. The lack of a resonance at δ = 5.00–7.00 ppm associated with the vinyl protons denoted complete conversion of the terminal double bonds during the Michael addition reaction and the presence of a peak at δ = 8.27 ppm indicated a protonated primary amine at the chain terminus. However, it is possible to notice in Fig. S4 (ESI†) that the relative intensities of the signals of the proton adjacent to the di-sulphide bond (yellow circle) and the protons in proximity of the newly formed secondary amine (pink circle) changes. This change was not proportional to the variation in feed ratio of ethylene-dioxy-bis-ethylamine and could be explained by hydrolysis of the ester bond on inter/intra molecular amidation reaction occurring between NH2 and ester bond in line with 13C and 2D-NMR spectroscopy.
Due to the intrinsic chemical similarity in terms of functionalities, the three OBAEs showed essentially superimposable ATR-IR spectra as evidenced in Fig. S6 (ESI†). FT-ATR-IR spectra of the OBAEs showed a broad signal at 3250 cm−1, typical of the stretching resonance of an extended H-bonded network of both secondary and primary amine groups, in contrast to the two sharp peaks at 3400 cm−1 and 3200 cm−1 in the spectrum of ethylene-dioxy-bis-ethylamine starting material due to primary amine functionality. The formation of a secondary amine was also confirmed by the shift of the N–H bending resonance from 1595 cm−1 to 1551 cm−1. The frequency of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band moved from 1725 cm−1 in the spectrum of DSD to 1643 cm−1 in the spectrum of OBAEs, as a result of the Michael addition reaction. Additionally, as for DSD, OBAEs showed a weak transition at around 670 cm−1 characteristics of C–S stretching.
O stretching band moved from 1725 cm−1 in the spectrum of DSD to 1643 cm−1 in the spectrum of OBAEs, as a result of the Michael addition reaction. Additionally, as for DSD, OBAEs showed a weak transition at around 670 cm−1 characteristics of C–S stretching.
ESI mass spectroscopy suggested molar masses ranging from 560 Da (OBAE C, Fig. S5C, ESI†) to 1434 Da (OBAE B, Fig. S5B, ESI†) and 2331 Da (OBAE A, Fig. S5A, ESI†) for the OBAEs synthesized, as expected from a step growth polymerization under the conditions employed. GPC data were difficult to interpret, perhaps owing to adsorption of the oligomers to the columns used, and thus additional characterisation methods were required. The number of reactive primary amines on the OBAEs were determined by fluorescamine assays, and the total number of basic amines was determined by acid–base titration (Fig. S7, ESI†). The values for the amine content of the oligomers were A: 0.85 mmol g−1; B 1.39 mmol g−1 and C: 3.27 mmol g−1. These results were in excellent agreement with fluorescamine assay data for OBAE C (also 3.27 mmol g−1), but less well-correlated for A and B, most likely due to their lower overall amine content.
Taken together, these data suggested a series of different materials, for which the theoretical structures derived from the most common fragments detected in mass spectrometry are shown in Fig. 3.
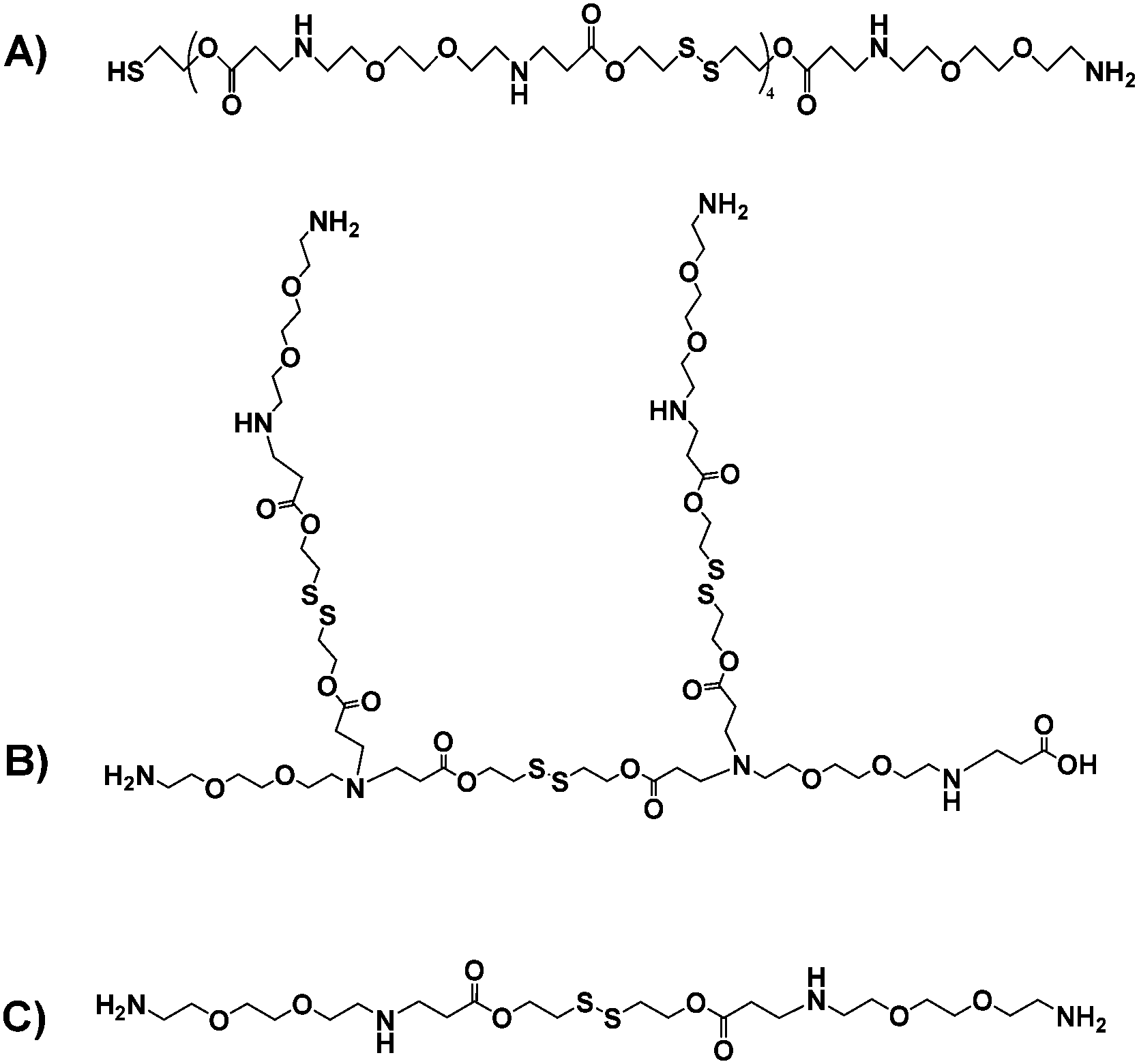 | ||
| Fig. 3 Putative structures of the OBAEs A, B and C based on the most common fragments from mass spectrometry and amine titration data. | ||
Prior papers describing poly(beta-aminoesters) have suggested structures deduced from the molar ratios of the functional groups of the monomers used, and have not always taken into account the potential formation of branches in the polymeric structure, even if diacrylate/diamine monomers have been used.28,29 In our case, we aimed for a variety of structures, including possible branching, such that the oligomers might condense with siRNA in different architectures during the ink-jet printing process.
We next explored the possibility to inkjet print the oligomers with siRNA. This method has been exploited for biomaterials and drug discovery,30–32 cell based therapies33 and for screening amorphous solid dispersions11 but to date only a few examples have been demonstrated for the formulation of micro- and nano-drug delivery systems.34–36 We thus screened the capability of the OBAEs to condense with siRNA by ink-jet printing in a 96-well plate starting from aqueous stock solutions of polymers and siRNA at different concentrations (Fig. 4). The amount of siRNA used was minimised by adoption of this method. For example, it was possible to perform 100 different experiments, with nine repeats of each OBAE/siRNA polyplex formulation (at each explored ratio), with as little as 800 ng of siRNA. The use of diluted solution of siRNA (0.01% w/v) allowed to work with ink formulations presenting low viscosity, close to the one of pure water (video as supporting data). By handling inks with such low viscosity, well-defined droplets were produced. Consequently, it has been simple to prevent the unwanted production of satellite droplets, as showed in Fig. S9 (ESI†).
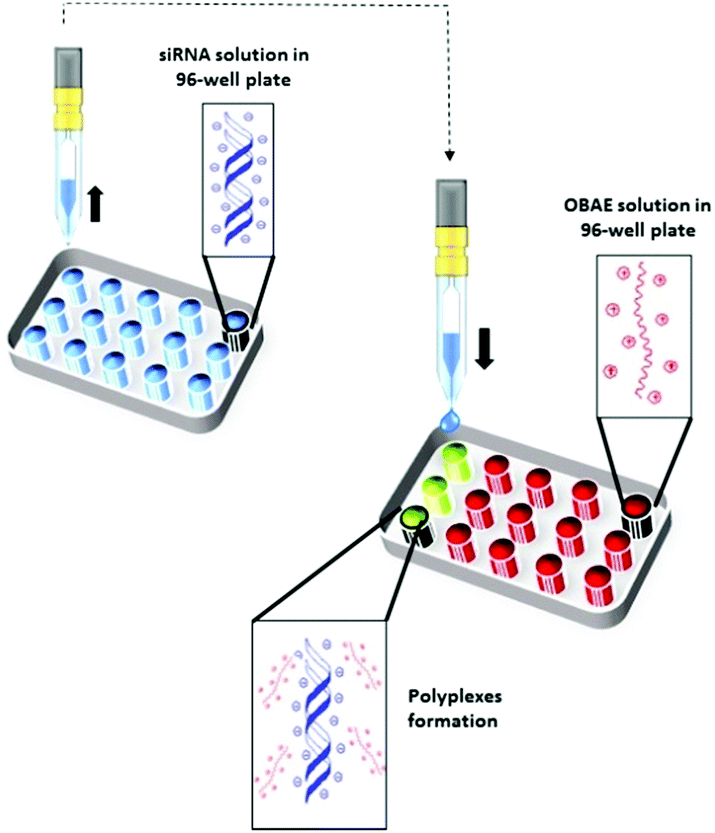 | ||
| Fig. 4 Schematic illustration of OBAE/siRNA polyplexes formation through ink-jet printing technique. | ||
The full set of polyplexes was prepared in less than 20 min by adopting a fully automated loop, and with the further advantage of storing all the samples in the compact space of a single 96-wellplate. The properties of the obtained polyplexes were compared with those made by conventional manual nanoprecipitation routes in order to confirm the potential and the reproducibility of this technique in the formation of drug delivery systems (Fig. S8A and B, ESI†). Polyplexes at different polymer/siRNA ratios were characterized in terms of size and polydispersity index through dynamic light scattering (DLS) (Fig. 5A). AFM analysis were also carried out, as shown for the OBAE C/siRNA polyplex at polymer/siRNA 10:1 weight ratio to better clarify the morphology, size and shape of the polyplexes. As evident in Fig. 5B, the OBAE/siRNA polyplexes were of spherical shape with size distributions in line with DLS data (between 200 nm and 500 nm).
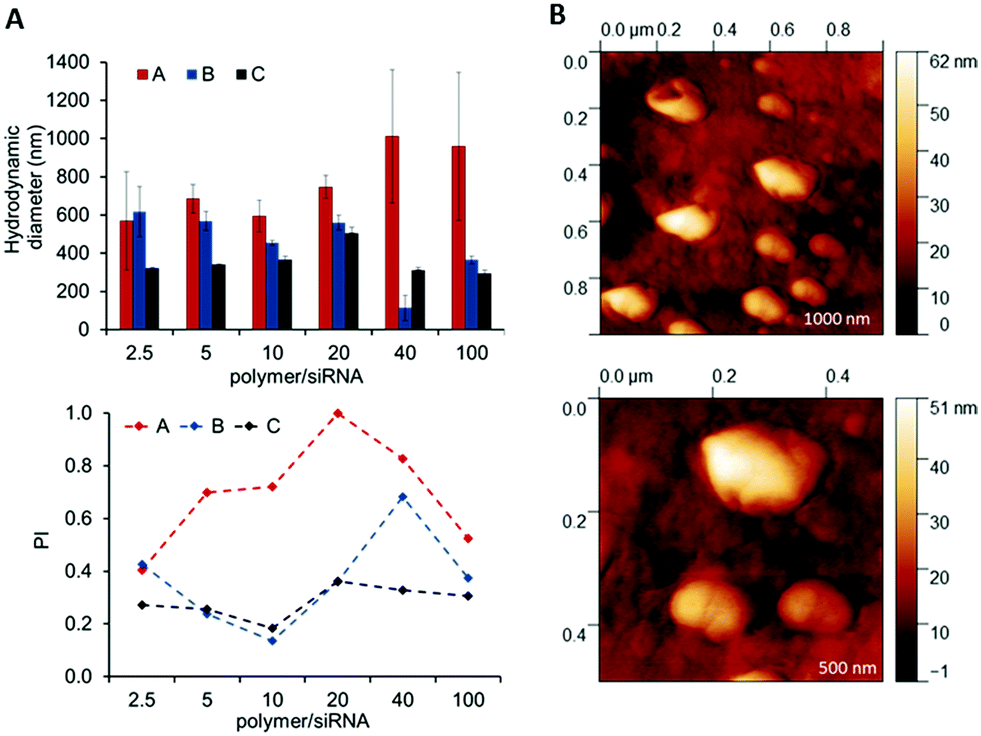 | ||
| Fig. 5 Characterization of OBAE/siRNA polyplexes. (A) Size and polydispersity index of OBAE/siRNA polyplexes at different polymer/siRNA weight ratios. (B) AFM images of OBAE C/siRNA polyplex at polymer/siRNA 10:1 weight ratio. | ||
The OBAEs were found to exhibit different behaviours in terms of complexation with siRNA, as expected from their chemical structures and charge densities. In particular, OBAE A formed polyplexes ranging from 600 to 1000 nm characterized by high polydispersity indexes, depending on the amount of siRNA condensed. In contrast, OBAE C showed the best performance in terms of complexation, thus forming polyplexes from 200 to 450 nm with an uniform size distribution in all the siRNA concentrations tested.
The ability of the OBAEs to form complexes with siRNA was investigated by agarose gel retardation (Fig. 6A) and ethidium bromide displacement assays (Fig. 6B). For the latter experiment, calf thymus DNA was incubated with ethidium bromide for 30 min and thereafter mixed with polyplexes at different polymer/siRNA ratio. Ethidium bromide displacement was monitored by fluorescence spectroscopy. In addition, experiments were carried out to simulate the behaviour of the polyplexes in an intracellular reducing environment. Accordingly, polyplexes were dispersed in buffer solutions containing GSH (10 mM) and their size was monitored throughout 2 h of incubation. As apparent from Fig. 6C, a marked change in the median diameters of particles in solution from ∼100 nm to ∼10 nm was observed after addition of GSH, indicating disassembly of the polyplexes following reductive stimulus.
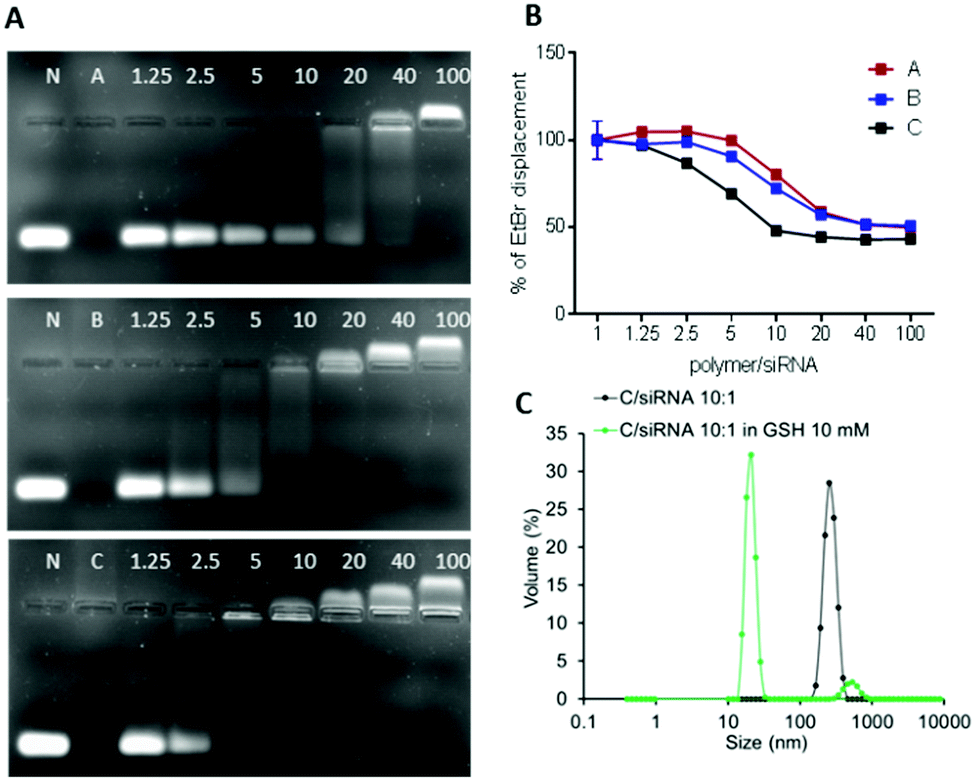 | ||
| Fig. 6 (A) siRNA condensation by A, B and C at different polymer/siRNA weight ratios as evaluated by the gel retardation assay. N represents naked siRNA. (B) Ethidium bromide displacement assay. (C) Stability of OBAE C/siRNA polyplex at polymer/siRNA 10:1 weight ratios incubated in GSH 10 mM for 2 h. | ||
The obtained results combined with the gel retention assays, confirmed the different condensation capabilities of OBAEs with siRNA, in line with their different charge densities. In particular, it was evident from the dye displacement and gel retardation assays that the siRNA binding affinity per unit mass of OBAEs increased from sample A to sample C, and that the polyplexes were disassembled in reducing environments. No significant difference in siRNA condensation capacity of OBAEs via jet printing or manual method was found (Fig. S8C, ESI†).
The biological effects of the OBAE/siRNA polyplexes were investigated in A549 lung cancer cells. In this study we designed disulfide-linked OBAEs of intermediate molar mass such that the oligomers would have sufficient charge to associate with siRNA during transit across cellular barriers, but also have an ability to depolymerise rapidly in the reducing intracellular environments to fragments which would have low affinity for siRNA and also low cytotoxicity.16,37,38 We therefore compared the effects of free OBAEs on the metabolic activities of A549 cells to those of the widely-used transfection agent branched PEI (25 kDa) after 4 h of treatment (Fig. 7A). Gene knockdown was then evaluated in an A549 cell line which constitutively expressed luciferase, using an anti-luciferase siRNA (CCGCAAGAUCCGCGAGAUU) and a control siRNA with a non-coding (scrambled) sequence. Cells were incubated for 4 h with OBAE/siRNA polyplexes at OBAE/siRNA 10:1 weight ratio (10 μg mL−1 of polyplexes) (Fig. 7B).
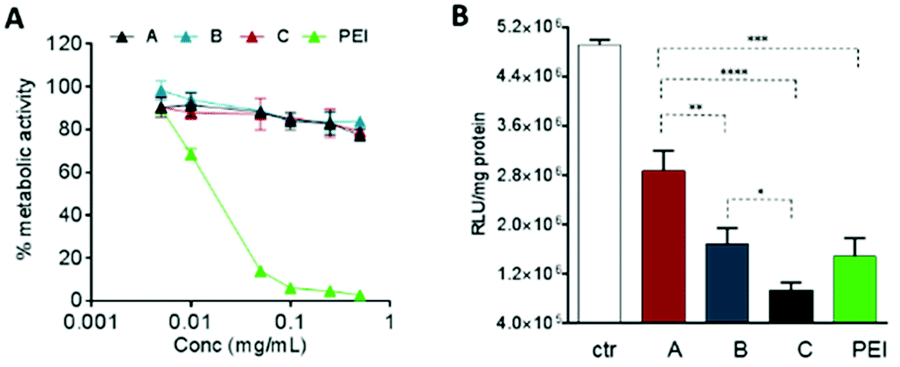 | ||
| Fig. 7 (A) Cytotoxicity of free OBAEs and (B) in vitro luciferase siRNA transfection from polyplexes vs. free siRNA (ctr) in A549-luciferase expressing cells after 4 h of incubation. RLU = relative light units, a measure for luciferase expression. Results are expressed as mean ± SD of three experiments. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05 two-way ANOVA test. | ||
The A549 cells retained ∼80% metabolic activity even when treated with the higher concentration of OBAEs and were significantly less toxic compared to PEI, which caused cell death at similar concentrations. As expected from the different siRNA binding properties of the OBAEs developed, a progressive increase in transfection efficiency was observed from oligomer A to oligomer C, with an overall knockdown activity of OBAE C greater than that of PEI at the same weight ratio (Fig. 7B), independently by the preparation method (Fig. S8D, ESI†). The intracellular transport of the polyplexes was probed in preliminary confocal microscopy experiments using a fluorescent Cy™3-tagged siRNA (Fig. 8). Inspection of the micrographs indicated that a progressive increase in siRNA internalization occurred ranging from oligomer A to oligomer C at the same concentration and time frame, in line with the expected trend based on the transfection results.
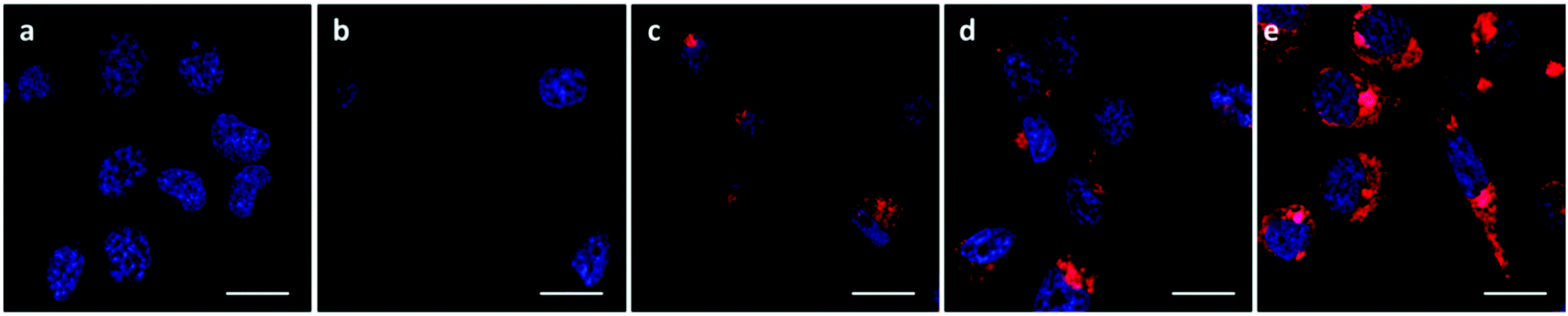 | ||
| Fig. 8 Confocal images of A549-luciferase expressing cells after 4 h of incubation with 10 μg mL−1 of polyplexes. (A) Uptake of OBAE/Cy3-siRNA polyplexes at 10:1 polymer/siRNA weight ratio. Cell nuclei were stained with DAPI (blue) and the images were acquired with 545 nm excitation and LP 560 nm spectral filters for Cy3-siRNA detection (red). (a) untreated cells, (b) naked siRNA, (c) OBAE A polyplexes, (d) OBAE B polyplexes, (e) OBAE C polyplexes. Zen 2009 image Software was utilized for image processing. Scale bar: 20 μm. | ||
The successful knockdown indicated that some of the polyplexes were able to escape to the reducing cytosolic regions where oligomer breakdown enabled delivery of the siRNA. Based on the previously described titration curves, the buffering capacities of the polymers were calculated to be 17, 24 and 56% for OBAE A, B and C respectively. Thus it was expected that OBAE C might be the most effective as an endosomal buffering agent to exploit the ‘proton sponge’ effect. As apparent from Fig. 9, a partial co-localization of the delivered siRNA with the lysosomes was found for OBAE C, suggesting that these complexes were initially trafficked to endolysosomal compartments. The subsequent enhanced knockdown achieved by these complexes was indicative that the OBAE C polyplexes were more stable in these regions compared to those of A and B, and were hence able to deliver siRNA more effectively following endo-lysosomal escape.
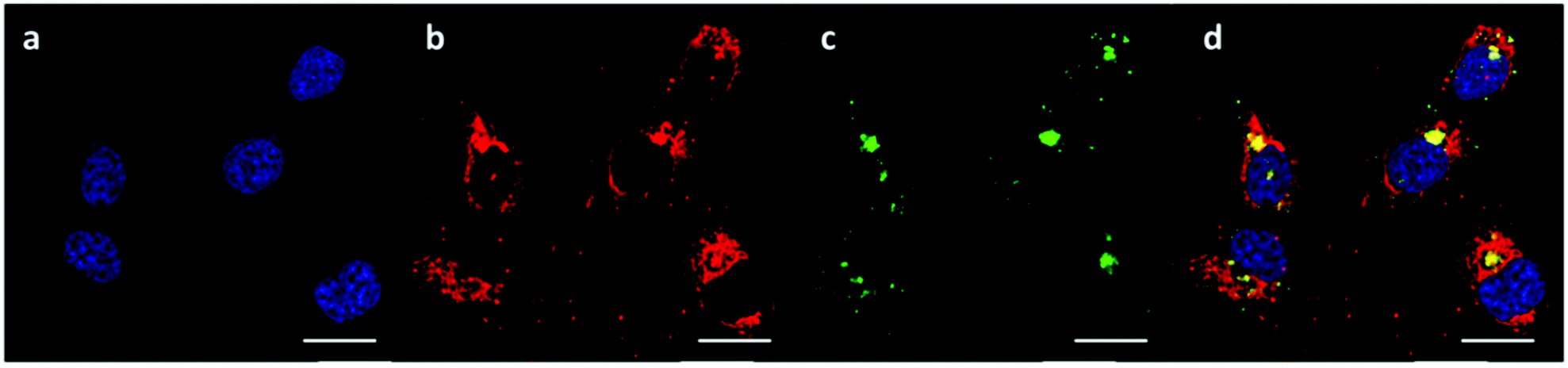 | ||
| Fig. 9 Intracellular trafficking of OBAE C/Cy3-siRNA polyplexes at 10:1 polymer/siRNA weight ratio. (a) Nucleus stained with DAPI, (b) Cy3-siRNA emission (ex: 555 nm excitation, em: LP 560), (c) Lysotracker Green DND-26 emission (ex: 488 nm, em: SP 555), (d) merge. Zen 2009 image Software was utilized for image processing. Scale bar: 20 μm. | ||
The data together indicated that the lowest molar mass OBAE was the most effective in terms of ease of formulation via ink-jet printing, and also in delivering siRNA to knock down the activity of luciferase. Based on NMR and mass spectrometry data, OBAE C was identified as the adduct of 2 ethylene-dioxy-bis-ethylamine monomers bridged by a single DSD unit, and would therefore have the highest number of basic amines per unit mass of the three OBAEs prepared. The titration data confirmed this assertion, and it was thus not surprising that OBAE C was the most effective of the candidates in siRNA condensation and polyplex formation. Our aim in this study was not to optimise the delivery systems, but to identify early in a synthesis/formulation cycle which, out of a pool of potential nucleic acid carriers, might be best from a printing and primary efficacy perspective. The fact that we were able to identify rapidly an oligo-beta-amino ester, which was as active as PEI in transfection but with much reduced effects on metabolic activity, using this method is indicative of its promise for future synthesis and formulation strategies.
Conclusions
Here we have shown the synthesis and characterisation of redox responsive oligo-β-aminoesters for siRNA delivery into cancer cells. Through an appropriate modulation of the molar ratio of the starting materials, we obtained oligo-β-aminoesters with different structures and charge densities. These cationic materials were rapidly condensed with siRNA through a facile and versatile high-throughput inkjet method, thus forming polyplexes with different siRNA binding affinities as well as colloidal properties. The new OBAEs showed reduced toxicity against cancer cells compared to the standard comparator PEI, yet retained a comparable high efficacy for transfection of siRNA in A549 cells. Future work will investigate the in vivo activities of these promising materials.Data access statement
All raw data created during this research are openly available from the corresponding authors cameron.alexander@nottingham.ac.uk and claudia.conte@unina.it and at the University of Nottingham Research Data Management Repository (https://rdmc.nottingham.ac.uk/). All analyzed data supporting this study are provided as ESI† accompanying this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Claudia Conte was supported by a fellowship by Associazione Italiana per la Ricerca sul Cancro (AIRC) co-funded by the European Union (iCARE/Marie Curie 2014). Tatiana Lovato thanks the EPSRC for a Doctoral Prize Fellowship. This work was supported by the Engineering and Physical Sciences Research Council grant numbers EP/H005625/1, EP/N03371X/1 and a Royal Society Wolfson Research Merit Award (WM150086) to CA. We also thank Tom Booth, Paul Cooling, Esme Ireson and Christine Grainger-Boultby for technical support.References
- Z. Zhou, X. Liu, D. Zhu, Y. Wang, Z. Zhang, X. Zhou, N. Qiu, X. Chen and Y. Shen, Adv. Drug Delivery Rev., 2017, 115, 115–154 CrossRef CAS PubMed.
- A. N. Zelikin, C. Ehrhardt and A. M. Healy, Nat. Chem., 2016, 8, 997–1007 CrossRef CAS PubMed.
- M. A. Islam, E. K. G. Reesor, Y. Xu, H. R. Zope, B. R. Zetter and J. Shi, Biomater. Sci., 2015, 3, 1519–1533 RSC.
- A. Samanta and I. L. Medintz, Nanoscale, 2016, 8, 9037–9095 RSC.
- D. W. Bartlett and M. E. Davis, Bioconjugate Chem., 2007, 18, 456–468 CrossRef CAS PubMed.
- J. C. Kasper, D. Schaffert, M. Ogris, E. Wagner and W. Friess, J. Controlled Release, 2011, 151, 246–255 CrossRef CAS PubMed.
- S. C. Semple, A. Akinc, J. X. Chen, A. P. Sandhu, B. L. Mui, C. K. Cho, D. W. Y. Sah, D. Stebbing, E. J. Crosley, E. Yaworski, I. M. Hafez, J. R. Dorkin, J. Qin, K. Lam, K. G. Rajeev, K. F. Wong, L. B. Jeffs, L. Nechev, M. L. Eisenhardt, M. Jayaraman, M. Kazem, M. A. Maier, M. Srinivasulu, M. J. Weinstein, Q. M. Chen, R. Alvarez, S. A. Barros, S. De, S. K. Klimuk, T. Borland, V. Kosovrasti, W. L. Cantley, Y. K. Tam, M. Manoharan, M. A. Ciufolini, M. A. Tracy, A. de Fougerolles, I. MacLachlan, P. R. Cullis, T. D. Madden and M. J. Hope, Nat. Biotechnol., 2010, 28, 172–176 CrossRef CAS PubMed.
- A. L. Troutier-Thuilliez, J. Thevenot, T. Delair and C. Ladaviere, Soft Matter, 2009, 5, 4739–4747 RSC.
- M. Sanna, G. Sicilia, A. Alazzo, N. Singh, F. Musumeci, S. Schenone, K. A. Spriggs, J. C. Burley, M. C. Garnett, V. Taresco and C. Alexander, ACS Med. Chem. Lett., 2018, 9, 193–197 CrossRef CAS PubMed.
- I. Louzao, B. Koch, V. Taresco, L. Ruiz-Cantu, D. J. Irvine, C. J. Roberts, C. Tuck, C. Alexander, R. Hague, R. Wildman and M. R. Alexander, ACS Appl. Mater. Interfaces, 2018, 10, 6841–6848 CrossRef CAS PubMed.
- V. Taresco, I. Louzao, D. Scurr, J. Booth, K. Treacher, J. McCabe, E. Turpin, C. A. Laughton, C. Alexander, J. C. Burley and M. C. Garnett, Mol. Pharmaceutics, 2017, 14, 2079–2087 CrossRef CAS PubMed.
- P. Mastorakos, C. Zhang, E. Song, Y. E. Kim, H. W. Park, S. Berry, W. K. Choi, J. Hanes and J. S. Suk, J. Controlled Release, 2017, 262, 37–46 CrossRef CAS PubMed.
- S. Y. Tzeng, D. R. Wilson, S. K. Hansen, A. Quinones-Hinojosa and J. J. Green, Bioeng. Transl. Med., 2016, 1, 149–159 CAS.
- D. G. Anderson, D. M. Lynn and R. Langer, Angew. Chem., 2003, 42, 3153–3158 CrossRef CAS PubMed.
- C. G. Zamboni, K. L. Kozielski, H. J. Vaughan, M. M. Nakata, J. Kim, L. J. Higgins, M. G. Pomper and J. J. Green, J. Controlled Release, 2017, 263, 18–28 CrossRef CAS PubMed.
- A. A. Eltoukhy, D. J. Siegwart, C. A. Alabi, J. S. Rajan, R. Langer and D. G. Anderson, Biomaterials, 2012, 33, 3594–3603 CrossRef CAS PubMed.
- S. Werth, B. Urban-Klein, L. Dai, S. Hobel, M. Grzelinski, U. Bakowsky, F. Czubayko and A. Aigner, J. Controlled Release, 2006, 112, 257–270 CrossRef CAS PubMed.
- J. C. Sunshine, D. Y. Peng and J. J. Green, Mol. Pharmaceutics, 2012, 9, 3375–3383 CrossRef CAS PubMed.
- F. Martello, M. Piest, J. F. Engbersen and P. Ferruti, J. Controlled Release, 2012, 164, 372–379 CrossRef CAS PubMed.
- A. A. Y. Almulathanon, E. Ranucci, P. Ferruti, M. C. Garnett and C. Bosquillon, Pharm. Res., 2018, 35, 86 CrossRef PubMed.
- P. Y. Teo, C. Yang, J. L. Hedrick, A. C. Engler, D. J. Coady, S. Ghaem-Maghami, A. J. T. George and Y. Y. Yang, Biomaterials, 2013, 34, 7971–7979 CrossRef CAS PubMed.
- X. J. Deng, N. Zheng, Z. Y. Song, L. C. Yin and J. J. Cheng, Biomaterials, 2014, 35, 5006–5015 CrossRef CAS PubMed.
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657–663 CrossRef CAS PubMed.
- A. Akinc and R. Langer, Biotechnol. Bioeng., 2002, 78, 503–508 CrossRef CAS PubMed.
- S. Y. Tzeng, B. P. Hung, W. L. Grayson and J. J. Green, Biomaterials, 2012, 33, 8142–8151 CrossRef CAS PubMed.
- K. L. Kozielski, S. Y. Tzeng and J. J. Green, Chem. Commun., 2013, 49, 5319–5321 RSC.
- C. Y. Hong, Y. Z. You, D. C. Wu, Y. Liu and C. Y. Pan, J. Am. Chem. Soc., 2007, 129, 5354–5355 CrossRef CAS PubMed.
- J. Kim, J. C. Sunshine and J. J. Green, Bioconjugate Chem., 2014, 25, 43–51 CrossRef CAS PubMed.
- A. A. Eltoukhy, D. Chen, C. A. Alabi, R. Langer and D. G. Anderson, Adv. Mater., 2013, 25, 1487–1493 CrossRef CAS PubMed.
- D. G. Anderson, S. Levenberg and R. Langer, Nat. Biotechnol., 2004, 22, 863–866 CrossRef CAS PubMed.
- E. A. Clark, M. R. Alexander, D. J. Irvine, C. J. Roberts, M. J. Wallacec, S. Sharpe, J. Yoo, R. J. M. Hague, C. J. Tuck and R. D. Wildman, Int. J. Pharm., 2017, 529, 523–530 CrossRef CAS PubMed.
- A. B. M. Buanz, C. C. Belaunde, N. Soutari, C. Tuleu, M. O. Gul and S. Gaisford, Int. J. Pharm., 2015, 494, 611–618 CrossRef CAS PubMed.
- T. Xu, J. Rohozinski, W. X. Zhao, E. C. Moorefield, A. Atala and J. J. Yoo, Tissue Eng., Part A, 2009, 15, 95–101 CrossRef CAS PubMed.
- T. Ehtezazi, N. M. Dempster, G. D. Martin, S. D. Hoath and I. M. Hutchings, J. Pharm. Sci., 2014, 103, 3733–3742 CrossRef CAS PubMed.
- S. Hauschild, U. Lipprandt, A. Rumplecker, U. Borchert, A. Rank, R. Schubert and S. Forster, Small, 2005, 1, 1177–1180 CrossRef CAS PubMed.
- N. Scoutaris, S. Ross and D. Douroumis, Pharm. Res., 2016, 33, 1799–1816 CrossRef CAS PubMed.
- H. Y. Xue, S. M. Liu and H. L. Wong, Nanomedicine, 2014, 9, 295–312 CrossRef CAS PubMed.
- M. Breunig, U. Lungwitz, R. Liebl and A. Goepferich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14454–14459 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb01215f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |