
Design and fabrication of reduction-sensitive cell penetrating nanofibers for enhanced drug efficacy†
He
Dong  *ab
*ab
*ab
Abstract
This work demonstrates a modular design strategy based on the supramolecular assembly of multidomain peptides to fabricate reduction-responsive cell penetrating nanofibers (CPNs), which hold great promise for selective targeting of cancer therapeutics to tumor cells.
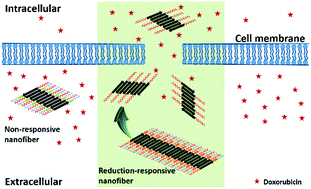
- This article is part of the themed collection: Journal of Materials Chemistry B Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

