
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoparticle-supported polymer brushes for temperature-regulated glycoprotein separation: investigation of structure–function relationship†
Huiting
Ma
 ab,
Lingdong
Jiang
a,
Solmaz
Hajizadeh
a,
Haiyue
Gong
a,
Bin
Lu
*b and
Lei
Ye
*a
ab,
Lingdong
Jiang
a,
Solmaz
Hajizadeh
a,
Haiyue
Gong
a,
Bin
Lu
*b and
Lei
Ye
*a
aDivision of Pure and Applied Biochemistry, Department of Chemistry, Lund University, Box 124, 221 00 Lund, Sweden. E-mail: lei.ye@tbiokem.lth.se
bKey Laboratory of Environment and Health, Ministry of Education & Ministry of Environmental Protection, State Key Laboratory of Environmental Health (Incubating), School of Public Health, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei 430030, China. E-mail: lubin@mails.tjmu.edu.cn
First published on 3rd May 2018
Abstract
In this work, we synthesized a series of nanoparticle-supported boronic acid polymer brushes for affinity separation of glycoproteins. Polymer brushes were prepared by surface-initiated atom transfer radical polymerization of glycidyl methacrylate and N-isopropylacrylamide, followed by stepwise modification of the pendant as well as the end functional groups to introduce boronic acid moieties through a Cu(I)-catalyzed alkyne–azide cycloaddition reaction. We investigated the impact of the polymer structure on glycoprotein binding under different pH and temperature conditions, and established new methods that allow glycoproteins to be more easily isolated and recovered with minimal alteration in solvent composition. Our experimental results suggest that for the separation of glycoproteins, terminal boronic acids located at the end of polymer chains play the most important role. The thermo-responsibility of the new affinity adsorbents, in addition to the high capacity for glycoprotein binding (120 mg ovalbumin per g adsorbent), provides a convenient means to realize simplified bioseparation not only for glycoproteins, but also for other carbohydrate-containing biological molecules.
Introduction
In recent years, polymer brushes have attracted much attention due to their unique structure and novel functional properties.1–4 Of particular interest is the grafting of macromolecules on the surfaces of different materials to improve their wettability, lubricity, hydrophilicity and colloidal stability.5,6 Among the different types of surface-initiated polymerization techniques, atom transfer radical polymerization (ATRP) is widely used because of its mild reaction conditions, versatile control of polymer structure and compatibility with a broad range of monomers.7–10 By using simple post-polymerization modification, polymer brushes made by ATRP can be easily converted into functional materials for use in bioseparation, enzyme catalysis, bioanalysis, etc.11,12Glycoproteins play an important role in many biological processes and are frequently used as biomarkers for different diseases. Effective separation of glycoproteins is one important step in many research areas and diagnostics.13–16 Lectin affinity chromatography (LAC) is widely used in this regard, as lectins can bind carbohydrates specifically. LAC has been realized using monolithic capillary columns, silica microparticles packed in microcolumns, sometimes hyphenated with mass spectrometry.17–20 However, LAC has some limitations such as being time-consuming process, and having complicated operations and high cost.21 Other methods e.g. based on capillary electrophoresis (CE) also use complicated protocols and require specialized instruments.22 Among all the methods used for glycoprotein enrichment, boronic acid-based affinity separation has become increasingly popular because of its low bias, long shelf time and convenient handling.23–28 In particular, polymer materials containing boronate affinity ligands can be designed to offer selective glycoprotein enrichment and separation, as boronic acids can form stable ester bonds with cis-diol groups at the carbohydrate site of glycoproteins. The sensitivity of boronate ester bonds towards pH variation can be used to modulate glycoprotein binding and release, thereby decreasing the influence of nonspecific protein binding.25,29 Different types of boronate affinity materials have been developed, for example, magnetic nanoparticles, monolithic capillary columns modified with boronic acids,30–32 and boronic acids immobilized on mesoporous silica33 as well as on agarose gel.34,35 Considering the steric hindrance and diffusion barrier encountered in protein separation, nanoparticles are ideal supports for immobilization of boronic acids to achieve efficient bioseparation.
In previous studies, we developed clickable boronic acids that could be easily conjugated to silica-supported polymer brushes for glycoprotein separation.13,36,37 The nanoparticle-supported polymer brushes displayed high capacity for glycoprotein binding, and the protein binding could be controlled by adjusting the solution temperature. In order to gain further insights into the structure–function relationship of nanoparticle-supported boronic acid polymer brushes, in this work we carried out a systematic investigation to study the impact of the polymer structure on glycoprotein binding. Glycidyl methacrylate (GMA), with and without N-isopropylacrylamide (NIPAm), was used as a monomer to prepare silica-supported polymer brushes by surface-initiated atom transfer radical polymerization (SI-ATRP). The copolymerization with NIPAm resulted in temperature-responsive polymer chains grafted on the nanoparticle surface. When the solution temperature was raised to above the lower critical solution temperature (LCST) of polyNIPAm (32 °C), the polymer brushes became dehydrated and collapsed on the surface,38,39 thereby affecting glycoprotein binding. The GMA monomer was used to introduce epoxide groups into the polymer brush that could be converted into pendant azide groups,40,41 which could be easily modified through a Cu(I)-catalyzed alkyne–azide cycloaddition reaction (click chemistry) to give the desired boronic acid polymer brushes.42 This work focused on studying the effect of the density and location of boronic acid ligands, and the influence of temperature on glycoprotein binding with the nanoparticle-supported polymer brushes. Besides studying the structure–function relationship, we were also interested in developing new methods for protein separation that require minimal alteration of the chemical composition of the protein solution, a condition more favourable for the final step in protein purification. For this purpose we intended to use the thermo-responsive properties of the polymer brushes to regulate the binding and release of glycoproteins.
Experimental
Materials
Tetraethylorthosilicate (TEOS), (3-aminopropyl)triethoxysilane (APTES), triethylamine (TEA), 2-bromoisobutyryl bromide (BIBB), N-isopropylacrylamide (NIPAm), glycidyl methacrylate (GMA), propargylamine, CuSO4, sodium ascorbate, sodium azide, N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PMDETA), tris[2-(dimethylamino)ethyl]amine (Me6TREN), Alizarin Red S (ARS), 3-aminophenylboronic acid hemisulfate (3-APBA hemisulfate), sodium hydrogen carbonate, propargyl chloroformate, ovalbumin (OVA), bovine serum albumin (BSA), horseradish peroxidase (HRP), sodium dodecyl sulfonate (SDS), sulphuric acid, acetonitrile (ACN), methanol, ammonia, toluene, N,N-dimethylformamide (DMF), tetrahydrofuran (THF), D-fructose, isopropanol and acetone were purchased from Sigma-Aldrich and were used without further purification. 2,2′-Azinobis[3-ethylbenzthiazoline-6-sulfonic acid]diammonium salt (1-Step ABTS) was purchased from Thermo Scientific. CuBr was stirred for 24 h in acetic acid, collected by centrifugation, washed with water and methanol, and dried in a vacuum before use. Ultrapure water (18.2 MΩ cm) was obtained from an ELGA LabWater System (Vivendi Water Systems Ltd). The clickable boronic acid, 3-(prop-2-ynyloxycarbonylamino)phenylboronic acid (PCAPBA), was synthesized according to our previously published procedure.36Preparation of nanoparticles and nanoparticle-supported polymer brushes
Using the same procedure, other boronic acid-modified polymer brushes, Si@pGMA-OH@BA, Si@pco-BA@BA, Si@pco-OH@BA and Si@pNIPAm@BA, were synthesized from Si@pGMA-OH@N3, Si@pco-N3@N3, Si@pco-OH@N3 and Si@pNIPAm@N3, respectively.
Characterization of nanoparticle-supported polymer brushes
Protein binding analysis
The amount of protein bound to the boronic acid-functionalized particles was investigated under three different conditions. The amount of protein binding was calculated using the equation:where C0 (mg mL−1) is the initial protein concentration, Ct (mg mL−1) is the concentration of the unbound protein, V (mL) is the volume of the protein solution, and m (in mg) is the mass of the composite particles. All the binding tests were carried out in triplicate, and the data are reported as mean value ± standard deviation.
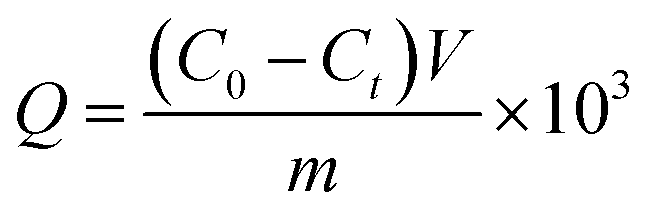
(1) In a series of 2 mL microcentrifuge tubes, 3 mg of boronic acid-functionalized particles was suspended in 1 mL of the protein solution. The mixture was shaken at 20 °C for 2 h, at a speed of 300 rpm using a thermal mixer. After centrifugation, 20 μL of the supernatant was taken to determine the free HRP by measuring the enzyme activity using 1-Step ABTS. Briefly, the 20 μL supernatant containing HRP was mixed with 150 μL of 1-Step ABTS in a microplate well. The microplate was incubated at 20 °C for 5 min, and then 100 μL of 1% SDS (in 20 mM PBS buffer, pH 9.0) was added to stop the enzymatic reaction. The absorbance of the reaction mixture at 410 nm was measured to calculate the amount of the unbound HRP.
(2) The remaining 980 μL mixture from step 1 (containing the nanoparticles and proteins) was heated to 40 °C and shaken at 300 rpm for 2 h. After centrifugation, 20 μL of the supernatant was taken to measure the amount of free HRP, using the procedure as described above.
(3) The remaining 960 μL mixture from step 2 was cooled to 20 °C before 1 mg of D-fructose was added. The mixture was shaken at 20 °C and at 300 rpm for 2 h. After centrifugation, 20 μL of the supernatant was taken to measure the amount of free HRP, using the procedure as described above.
The boronic acid-functionalized particles used were Si@pco-BA@BA, Si@pco-OH@BA, and Si@pNIPAm@BA.
(1) The composite particles were mixed with 1 mL of 0.2 M acetate buffer (pH 4.0) and shaken at 20 °C and at 300 rpm for 2 h. After centrifugation, 20 μL of the supernatant was taken to measure the amount of free HRP.
(2) The remaining 980 μL mixture from step 1 (containing the nanoparticles and proteins) was heated to 40 °C and shaken at 300 rpm for 2 h. After centrifugation, 20 μL of the supernatant was taken to measure the amount of free HRP.
(3) Similar to step 1, the initial composite particles with the bound glycoprotein were mixed with 1 mL of 0.2 M acetate buffer (pH 4.0) containing 1 mg mL−1D-fructose, and shaken at 20 °C and at 300 rpm for 2 h. After centrifugation, 20 μL of the supernatant was taken to measure the amount of free HRP.
(4) The remaining 980 μL mixture from step 3 (containing the nanoparticles and proteins) was heated to 40 °C and shaken at 300 rpm for 2 h. After centrifugation, 20 μL of the supernatant was taken to measure the amount of free HRP.
Results and discussion
Synthesis and characterization of nanoparticle-supported polymer brushes
In this work we designed a series of nanoparticle-supported polymer brushes functionalized with boronic acid at different locations in the polymer chains. The main idea was to find out the most suitable structure of nanoparticle-supported polymer brushes for glycoprotein separation. In addition to understanding this structure–function relationship, we were also interested in using thermodesorption to elute glycoproteins from the affinity materials, as this method might not involve the dramatic change in solvent conditions of protein samples, which can be beneficial for protein purification. The intermediate polymer brushes were synthesized from GMA and/or NIPAm according to our previous publications,13,36,37,42 and were further functionalized with the clickable boronic acid through a Cu(I)-catalyzed click reaction. The synthetic steps are illustrated in Scheme 1. Firstly, silica nanoparticles were prepared using the one-step Stöber process.43 Amino groups were then introduced onto the silica nanoparticles to allow the ATRP initiator to be immobilized. The initiator-functionalized nanoparticles, Si@initiator, were then used to prepare three types of nanoparticle-supported polymer brushes: Si@pGMA, Si@pco and Si@pNIPAm. In the next step, the epoxide rings in the GMA moieties were converted into either pendant azide or diol groups by treatment with NaN3 or sulfuric acid, and the terminal Br was converted into azide by reacting with NaN3. Finally, the azide groups in the polymer brushes were used to introduce the clickable boronic acid with the high efficiency click reaction.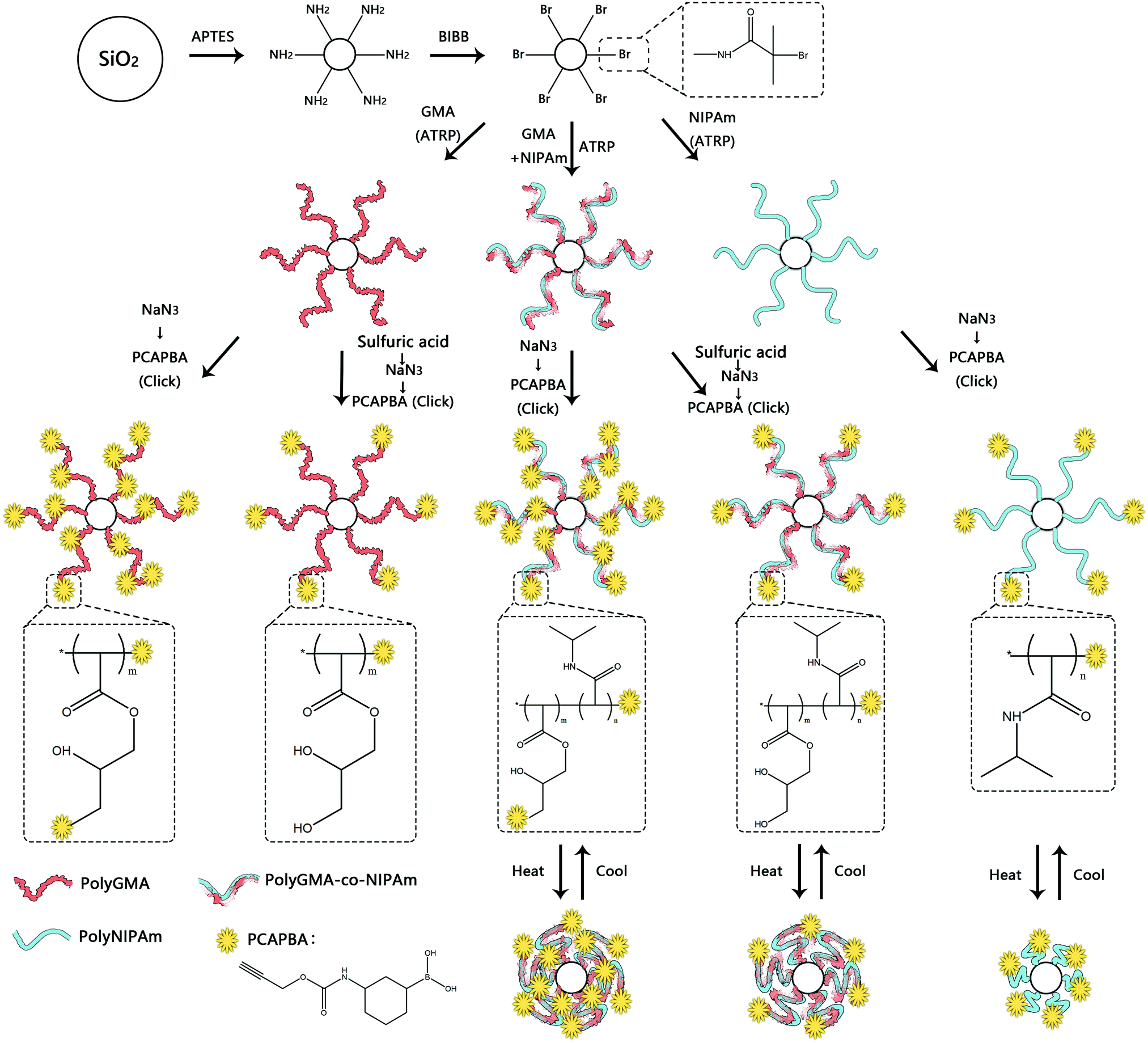 | ||
| Scheme 1 Synthesis of boronic acid-functionalized polymer brushes on silica nanoparticles. | ||
The nanoparticles synthesized in this work (including Si, Si@NH2, Si@initiator, and the azide-functionalized nanoparticles (Si@pGMA-N3@N3, Si@pGMA-OH@N3, Si@pco-N3@N3, Si@pco-OH@N3, Si@pNIPAm@N3)) were analyzed using SEM. As seen from the SEM images (Fig. 1 and Fig. S1, ESI†), the original Si nanoparticles were spherical with a smooth surface (Fig. 1a) and had a diameter of 100–150 nm. After grafting the polymer brushes on the surface, the obtained composite particles maintained a spherical shape, but the roughness of the particles increased obviously (Fig. 1b–f).
 | ||
| Fig. 1 SEM images of nanoparticles (a) Si, (b) Si@pGMA-N3@N3, (c) Si@pGMA-OH@N3, (d) Si@pco-N3@N3, (e) Si@pco-OH@N3, (f) Si@pNIPAm@N3. | ||
The chemical composition of the nanoparticles obtained after different synthetic steps was analysed by FT-IR. In Fig. 2, the most intense absorption peak appears at 1100 cm−1, characteristic of the Si–O–Si vibration band. The peak at 1720 cm−1 is attributed to the carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in Si@polyGMA (Fig. 2b). After treatment with sodium azide and conjugation with the clickable boronic acid, a residual azide signal at 2100 cm−1 was still observed from Si@pGMA-BA@BA (Fig. 2c), which could be explained as the unreacted azide groups remaining in the polymer brushes, possibly due to the steric hindrance encountered during the click reaction. In the case of Si@pGMA-OH@BA, no azide signal was found (Fig. 2d), which can be explained by the low abundance of azide in its precursor Si@pGMA-OH@N3, as well as complete conversion of the terminal azide into boronic acid. For Si@pco, in addition to the IR signals from the polymerized GMA monomer, new absorption bands appeared at 1650 cm−1 and 1540 cm−1, which are the characteristic amide signals due to the polymerized NIPAm (Fig. 2e). After Si@pco was reacted with sodium azide and conjugated to the clickable boronic acid, the product Si@pco-BA@BA also displayed a residual azide signal at 2100 cm−1 (Fig. 2f), similar to that observed from Si@pGMA-BA@BA (Fig. 2c), possibly due to the incomplete conversion of azide into boronic acid caused by the steric hindrance during the click reaction. Like for Si@pGMA-OH@BA, the low abundance azide in Si@pco-OH@BA could not be detected after the terminal azide was converted into boronic acid via the click reaction (Fig. 2g). From the two nanoparticle-supported polyNIPAm brushes, Si@pNIPAm and Si@pNIPAm@BA, the characteristic amide I and amide II bands at 1650 cm−1 and 1540 cm−1 are obvious but are relatively weak, suggesting a lower content of the organic polymer in these composite particles.
O) in Si@polyGMA (Fig. 2b). After treatment with sodium azide and conjugation with the clickable boronic acid, a residual azide signal at 2100 cm−1 was still observed from Si@pGMA-BA@BA (Fig. 2c), which could be explained as the unreacted azide groups remaining in the polymer brushes, possibly due to the steric hindrance encountered during the click reaction. In the case of Si@pGMA-OH@BA, no azide signal was found (Fig. 2d), which can be explained by the low abundance of azide in its precursor Si@pGMA-OH@N3, as well as complete conversion of the terminal azide into boronic acid. For Si@pco, in addition to the IR signals from the polymerized GMA monomer, new absorption bands appeared at 1650 cm−1 and 1540 cm−1, which are the characteristic amide signals due to the polymerized NIPAm (Fig. 2e). After Si@pco was reacted with sodium azide and conjugated to the clickable boronic acid, the product Si@pco-BA@BA also displayed a residual azide signal at 2100 cm−1 (Fig. 2f), similar to that observed from Si@pGMA-BA@BA (Fig. 2c), possibly due to the incomplete conversion of azide into boronic acid caused by the steric hindrance during the click reaction. Like for Si@pGMA-OH@BA, the low abundance azide in Si@pco-OH@BA could not be detected after the terminal azide was converted into boronic acid via the click reaction (Fig. 2g). From the two nanoparticle-supported polyNIPAm brushes, Si@pNIPAm and Si@pNIPAm@BA, the characteristic amide I and amide II bands at 1650 cm−1 and 1540 cm−1 are obvious but are relatively weak, suggesting a lower content of the organic polymer in these composite particles.
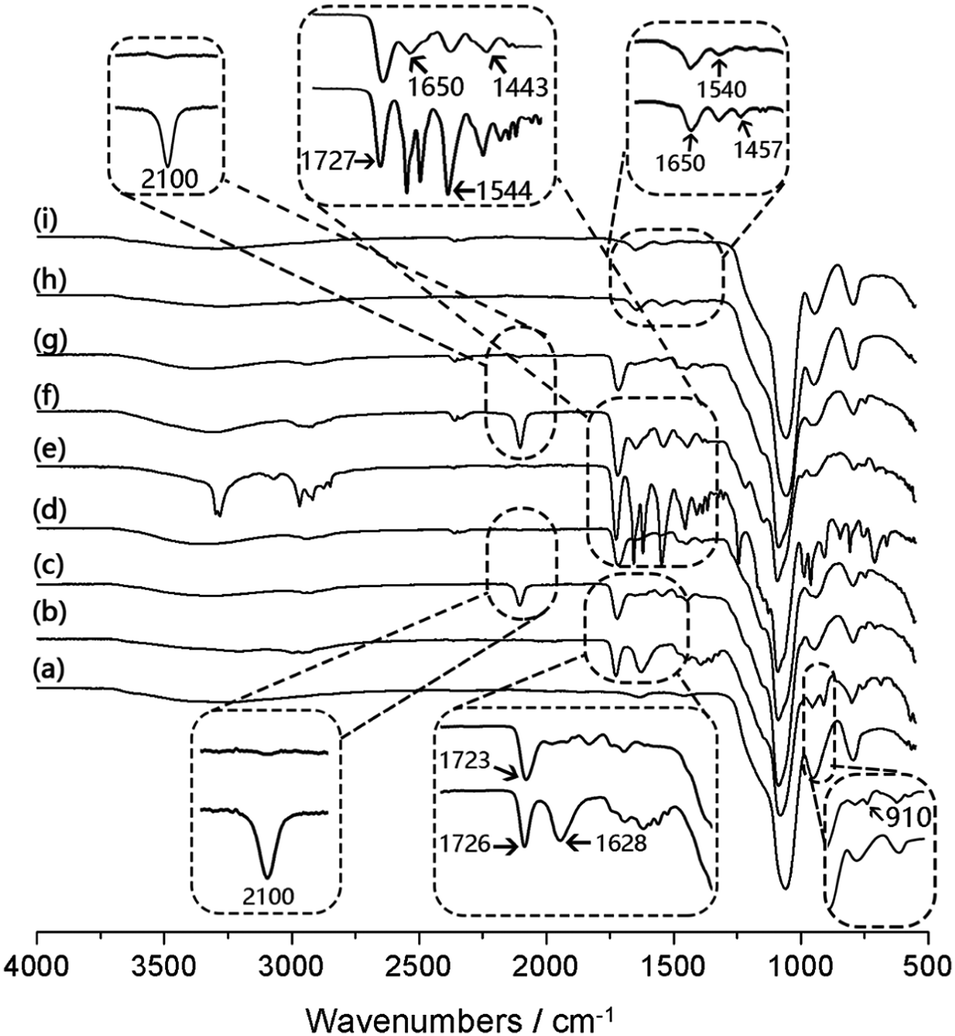 | ||
| Fig. 2 IR spectra of Si (a), Si@pGMA (b), Si@pGMA-BA@BA (c), Si@pGMA-OH@BA (d), Si@pco (e), Si@pco-BA@BA (f), Si@pco-OH@BA (g), Si@pNIPAm (h) and Si@pNIPAm@BA (i). | ||
The organic content of the different composite particles was assessed by TGA, and the chemical composition of the composite particles was measured by elemental analysis. These experimental results are presented in Table 1 and Fig. S3 (ESI†). In the TGA data, the weight loss under 200 °C is attributed to the evaporation of water. The residual weight remaining after the TGA was used to calculate the content of the inorganic silica material.
| Particles | C (%) | H (%) | N (%) | Br (%) | B (%) | Si (%) | B/Si (mmol g−1) | B/polymer (mmol g−1) |
|---|---|---|---|---|---|---|---|---|
| Si@NH2 | 0.21 | 1.49 | 1.22 | n.d. | n.d. | 94.0 | — | — |
| Si@initiator | 3.88 | 1.94 | 0.51 | 1.33 | n.d. | 87.5 | — | — |
| Si@pGMA | 35.16 | 5.26 | 1.33 | 0.12 | n.d. | 38.6 | — | — |
| Si@pGMA-N3@N3 | 34.7 | 5.15 | 12.58 | 0.02 | n.d. | 17.3 | — | — |
| Si@pGMA-BA@BA | 29.53 | 4.42 | 7.72 | n.d. | 0.34 | 42.0 | 0.75 | 0.54 |
| Si@pGMA-OH | 28.69 | 4.94 | 0.33 | 0.12 | n.d. | 37.8 | — | — |
| Si@pGMA-OH@N3 | 28.44 | 4.8 | 0.71 | 0.02 | n.d. | 40.7 | — | — |
| Si@pGMA-OH@BA | 32.00 | 5.27 | 0.91 | n.d. | 0.13 | 43.9 | 0.27 | 0.21 |
| Si@pco | 45.92 | 6.67 | 3.95 | 0.12 | n.d. | 28.3 | — | — |
| Si@pco-N3@N3 | 36.48 | 5.63 | 10.25 | 0.003 | n.d. | 24.1 | — | — |
| Si@pco-BA@BA | 20.27 | 5.36 | 4.99 | n.d. | 0.13 | 26.5 | 0.45 | 0.16 |
| Si@pco-OH | 34.02 | 5.93 | 3.04 | 0.11 | n.d. | 29.0 | — | — |
| Si@pco-OH@N3 | 31.7 | 5.08 | 2.08 | 0.002 | n.d. | 32.5 | — | — |
| Si@pco-OH@BA | 36.58 | 5.67 | 1.34 | n.d. | 0.08 | 29.5 | 0.25 | 0.1 |
| Si@pNIPAm | 7.59 | 2.39 | 1.49 | 0.13 | n.d. | 83.7 | — | — |
| Si@pNIPAm@N3 | 7.32 | 2.16 | 2.36 | 0.004 | n.d. | 73.2 | — | — |
| Si@pNIPAm@BA | 8.33 | 2.29 | 1.86 | n.d. | 0.09 | 82.1 | 0.10 | 0.5 |
Among all the boronic acid-functionalized composite particles, Si@pNIPAm@BA had the lowest organic content, which is in agreement with the weak amide signals in the IR spectra of the nanoparticle-supported polyNIPAm brushes, Si@pNIPAm and Si@pNIPAm@BA (Fig. 2h and i). In our previous work, we also observed that the efficiency of growing polyNIPAm by SI-ATRP is lower than that of polyGMA.
Based on the contents of boron and inorganic silica, the number of boronic acids conjugated to the silica-supported nanoparticle brushes was calculated. Thus the boronic acid-functionalized composite nanoparticles Si@pGMA-BA@BA, Si@pGMA-OH@BA, Si@pco-BA@BA, Si@pco-OH@BA and Si@pNIPAm@BA were found to contain 0.75, 0.27, 0.45, 0.25 and 0.10 mmol boronic acid per gram of silica, respectively. The variation of boronic acid contents in these materials was expected based on the abundance of azide groups in their precursor particles Si@pGMA-N3@N3, Si@pGMA-OH@N3, Si@pco-N3@N3, Si@pco-OH@N3 and Si@pNIPAm@N3.
The content of boron in the final polymer can be calculated from the elemental analysis and the TGA results (Table 1, the last column). The content of boron was controlled by varying the density of azide groups in the polymer brush. Briefly, treating the epoxy-containing polymer (e.g. Si@pGMA) with sulfuric acid before introducing the azide leads to a lower boron content. For other composite particles, the boron content can be further adjusted by varying the amount of NIPAm relative to GMA, as well as by controlling the length of the polymer chain (by virtue of ATRP).
To confirm the successful conjugation of boronic acid into the polymer brushes, Si@pGMA-BA@BA, Si@pGMA-OH@BA, Si@pco-BA@BA, Si@pco-OH@BA and Si@pNIPAm@BA were treated with a fluorogenic cis-diol ARS to measure the binding-induced fluorescence emission.13 As shown in Fig. 3, all the boronic acid-functionalized particles emitted strong fluorescence after the particles were mixed with ARS in a basic buffer.
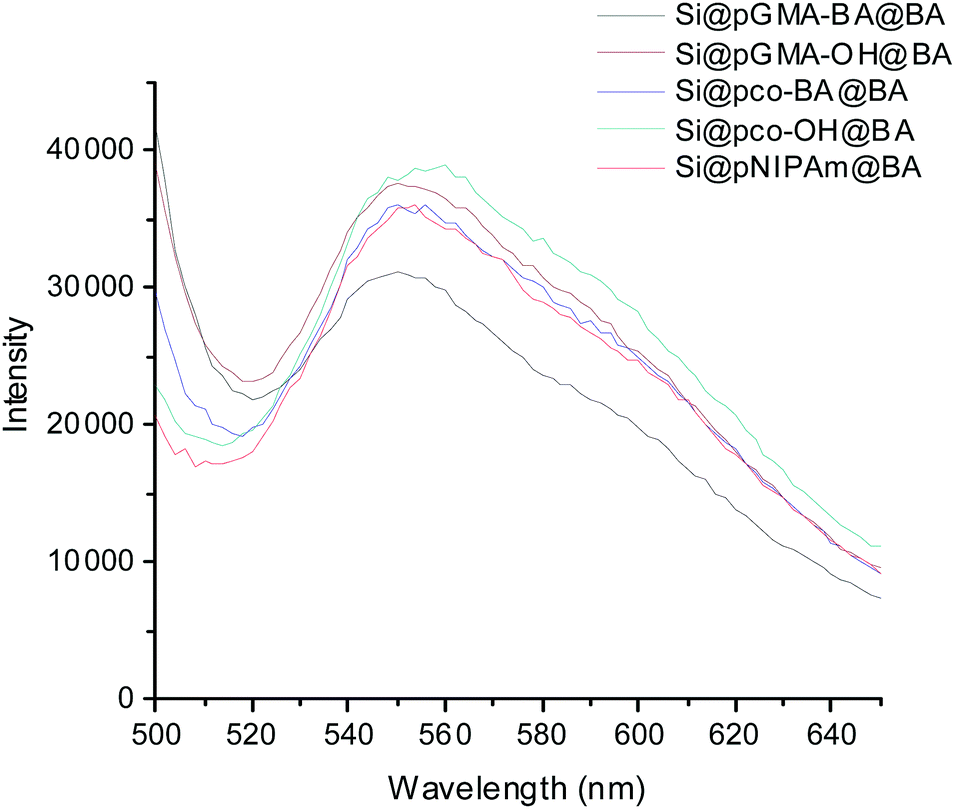 | ||
| Fig. 3 Fluorescence emission of boronic acid-functionalized composite particles after exposure to ARS. | ||
Binding of glycoproteins with different boronic acid-functionalized particles
To compare the protein-binding behaviours of the boronic acid-functionalized nanoparticles, we first carried out binding experiments in single protein solutions adjusted to different pH values and at different temperatures. OVA and HRP were used as model glycoproteins44,45 and BSA as a representative non-glycoprotein.46In order to compare the composite particles with different thermo-responsive properties more clearly, the equilibrium binding data in Fig. 4 are presented in two sets: one for the non-thermo-responsive particles Si@pGMA-BA@BA and Si@pGMA-OH@BA, and another for the thermo-responsive particles Si@pco-BA@BA, Si@pco-OH@BA and Si@pNIPAm@BA.
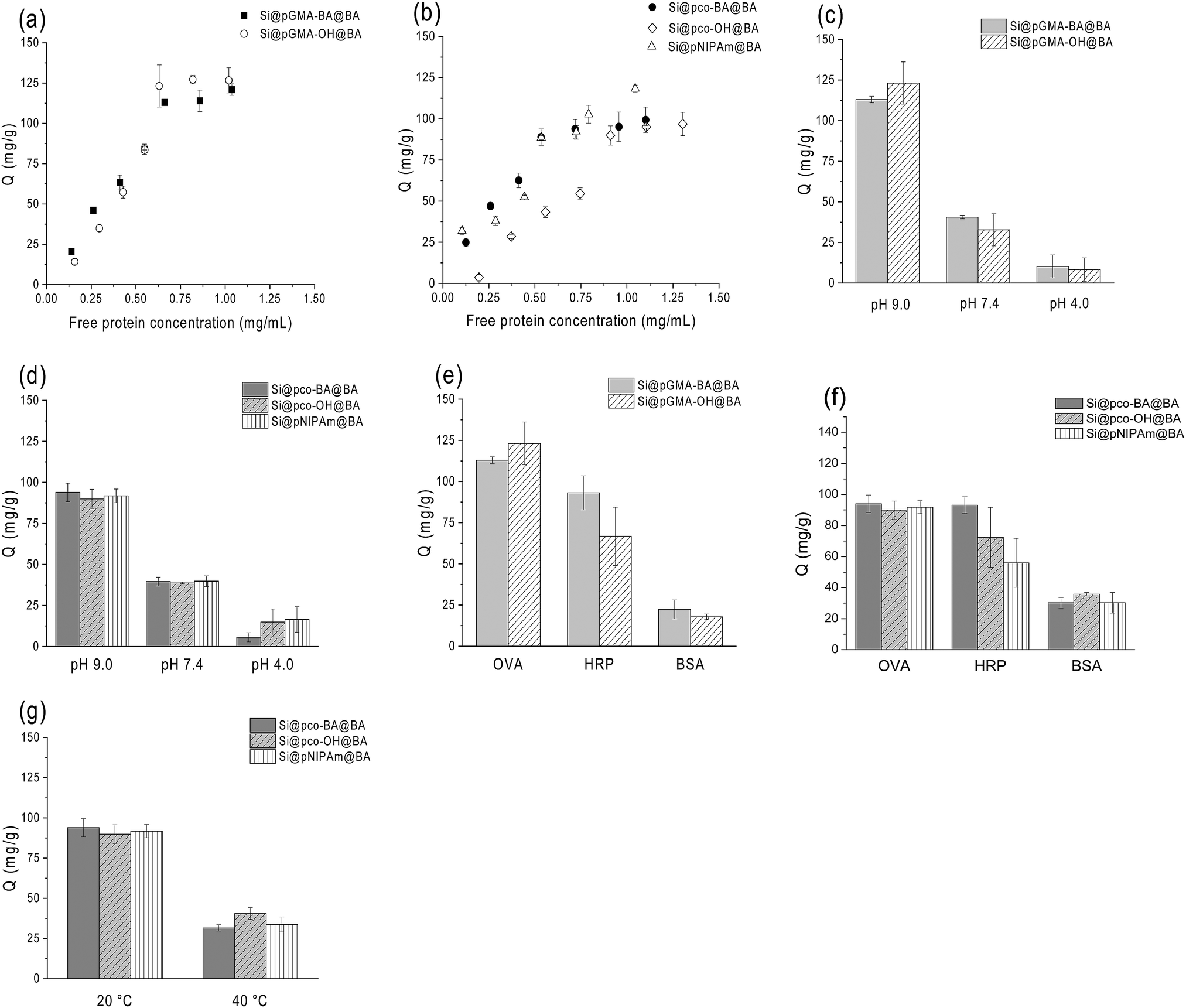 | ||
| Fig. 4 Protein binding characteristics. (a and b) Binding isotherms of OVA measured at 20 °C and pH 9.0. (c and d) OVA binding to the composite particles measured at 20 °C and different pH values. The initial concentration of OVA was 1 mg mL−1. (e and f) Binding of different proteins to the composite particles measured in pH 9 buffer at 20 °C. The initial concentration of the individual protein was 1 mg mL−1. (g) Binding of OVA to the thermo-responsive polymer brushes measured in pH 9 buffer at different temperatures. The initial OVA concentration was 1 mg mL−1. | ||
From Fig. 4a and b, it is clear that the boronic acid-functionalized polymer brushes without the NIPAm component had a higher capacity for OVA binding (∼125 mg g−1) than the thermo-responsive polymer brushers (∼100 mg g−1), both higher than the capacity reported in our previous work.37 The slightly higher capacity of Si@pGMA-BA@BA and Si@pGMA-OH@BA for glycoprotein binding can be explained as their higher boronic acid content than the thermo-sensitive copolymer brushes and polyNIPAm polymer brushes grafted on the silica nanoparticles (Si@pco-BA@BA, Si@pco-OH@BA and Si@pNIPAm@BA, Table 1). Interestingly, the polymer brushes functionalized with boronic acid at the chain termini displayed OVA capacity similar to that functionalized both at chain termini and at pendant side groups (i.e. Si@pGMA-OH@BA vs. Si@pGMA-BA@BA and Si@pco-OH@BA vs. Si@pco-BA@BA). It is likely that due to the high grafting density (as discussed in our previous work) and a steric effect, the terminal boronic acid played the most important role in binding the glycoprotein.
Glycoprotein binding with all the boronic acid-functionalized polymer brushes displayed clear pH-dependence. As seen in Fig. 4c and d, when the solution pH changed from 9 to 4, OVA binding to the composite particles decreased significantly. The pH-dependent OVA binding can be explained as the structural change of the appended phenylboronic acid upon pH variation. Under alkaline conditions the boron atom has a sp3 configuration and is able to form strong boronate ester bonds with cis-diols, whereas under acidic conditions the boron changes to a sp2 configuration, leading to hydrolysis of the boronate ester bonds to release the bound glycoprotein. The sensitivity of the boronate ester bond against pH can therefore be used to control the binding and dissociation of glycoproteins from boronate affinity materials.37,47
For the two types of glycoproteins tested, OVA and HRP, in general the uptake of OVA by the boronic acid-functionalized polymer brushes was higher than that of HRP (Fig. 4e and f). Under the same conditions the non-specific protein binding, as estimated from the BSA binding, was much lower. The difference of binding between OVA and HRP by boronate affinity materials has been observed in our previous investigations and can be explained as the content of the accessible cis-diol structures in these two glycoproteins. Although HRP has more glycosylation sites, OVA has a higher saccharide content and therefore can present more accessible cis-diols to bind the boronic acid polymer brushes.
To evaluate the water-compatibility of the nanoparticle-supported polymer brushes, we measured the rate of particle sedimentation in aqueous buffer by monitoring the change of optical transmittance of the particle suspension. As shown in Fig. S4a and b (ESI†), at 20 °C the particle Si@pGMA-BA@BA precipitated significantly faster than Si@pGMA-OH@BA. The faster sedimentation of Si@pGMA-BA@BA was due to the multiple phenylboronic acids in the polymer chains. The phenylboronic acids at low pH are relatively hydrophobic, making the composite particles form aggregates to sediment faster than at high pH. Because Si@pGMA-OH@BA contains abundant hydroxyl groups, the composite particle is hydrophilic in a wide pH range, and therefore it does not sediment even at low pH.
The three nanoparticle-supported polymer brushes containing NIPAm were designed to introduce thermo-responsive properties into the composite particles. In the literature it is well known that polyNIPAm has a lower critical solution temperature (LCST) of around 32 °C. Below the LCST polyNIPAm is hydrophilic and exists as an extended and hydrated chain, whereas above the LCST the polymer chain becomes hydrophobic and collapses. To verify that the two boronic acid-functionalized copolymer brushes have similar temperature-controlled structural changes, we studied the rate of particle sedimentation at 20 °C and 40 °C. As shown in Fig. S4c and d (ESI†), the two composite particles Si@pco-BA@BA and Si@pco-OH@BA dispersed in pH 9 buffer at 40 °C settled slightly faster than in the same buffer at 20 °C. The faster sedimentation can be explained as a result of the thermo-induced dehydration and collapse of the polymer brushes. We should note, however, that at 40 °C although the NIPAm components tend to make the polymer brushes collapse, the higher pH (pH 9) makes the boronic acids become negatively charged, leading to stabilizing the colloidal suspension. The overall result is that the sedimentation of Si@pco-BA@BA and Si@pco-OH@BA became not very obvious when the temperature increased from 20 °C to 40 °C.
One of our intriguing questions is if the temperature-controlled structural transition can be utilized to control the binding of glycoproteins to thermo-responsive polymer brushes. In the next experiment, we therefore studied the effect of solution temperature on the uptake of OVA by the three composite particles Si@pco-BA@BA, Si@pco-OH@BA and Si@pNIPAm@BA. As shown in Fig. 4g, when the solution temperature changed from 20 °C to 40 °C, for all the three polymer brushes the amount of OVA binding in pH 9 buffer decreased more than 60% (from ∼90 mg g−1 to ∼35 mg g−1). This profound temperature effect provides an interesting approach to modulate glycoprotein binding with the new boronate affinity particles, and should provide a convenient means to enable more simplified protein separation.
Verification of protein separation by SDS-PAGE
In order to observe more directly the protein separation with the temperature-responsive nanoparticles, we used SDS-PAGE analysis to monitor a mixture of OVA and BSA after it was treated with Si@pNIPAm@BA. The protein mixture was first incubated with Si@pNIPAm@BA at 20 °C, and then at 40 °C. The unbound proteins at these two different temperatures were analysed by SDS-PAGE.As shown in Fig. 5, at 20 °C both BSA (∼66 kDa) and OVA (∼45 kDa) were depleted from solution by the nanoparticles (Lane 2, 3). The small shift of the protein bands, compared to the marker (Lane 1), was caused by the difference in the buffer pH. The BSA binding can be attributed to nonspecific adsorption. When the temperature was raised to 40 °C, the majority of BSA was released from the nanoparticles, while the amount of OVA released was relatively less than BSA (Lane 4). The release of protein caused by the increased temperature can be explained as a result of the collapse of the polymer brushes on the particle surface, making the originally bound BSA be excluded from the composite particles. For OVA, because of its covalent binding to the terminal boronic acid, the effect of increased temperature is less than for BSA. After the washing steps, it is clear that the final protein solution eluted from the nanoparticles contained mainly OVA (Lane 6).
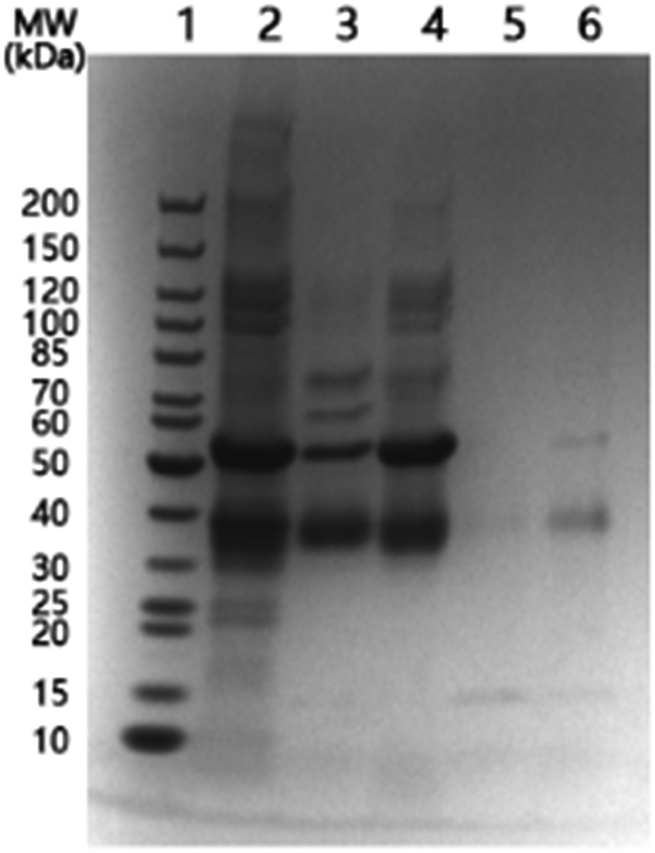 | ||
| Fig. 5 SDS-PAGE analysis of the protein sample treated with Si@pNIPAm@BA. Lane 1, protein marker; Lane 2, protein mixture containing 1 mg mL−1 OVA and 1 mg mL−1 BSA; Lane 3, unbound protein mixture at 20 °C; Lane 4, unbound protein mixture at 40 °C; Lane 5, collected washing buffer; and Lane 6, proteins eluted in acetate buffer (pH 4). | ||
Controlling glycoprotein binding via temperature and addition of monosaccharides
In conventional boronate affinity separation, variation of pH is the most used method to control the binding and release of cis-diols from boronic acid-functionalized adsorbents. Alternative methods to release glycoproteins at a constant pH, e.g. by adjusting temperature, can be useful because it does not require a significant adjustment of pH. As glycoprotein binding is mediated by the saccharide moieties, another way of releasing glycoprotein without changing the pH is by adding competing monosaccharides. As an example, under alkaline conditions D-fructose is able to form stable ester bonds with boronic acids, and it has been used as a competing cis-diol to displace glycoproteins from boronate affinity particles.42,48To investigate the effects of temperature and fructose on glycoprotein binding in a more realistic environment, we studied the binding of HRP on the three thermo-responsive particles in the presence of excess non-glycoprotein, BSA. In this experiment HRP was used as a model glycoprotein, as its concentration could be determined conveniently by measuring the enzyme activity. As shown in Fig. 6, all the three boronic acid-functionalized silica nanoparticles (containing NIPAm) showed a very clear response to temperature and D-fructose. It is noteworthy that temperature and fructose displayed a concerted effect on HRP binding: addition of fructose and increasing temperature resulted in the maximum release of HRP from the boronate affinity particles. The temperature and fructose response were not affected by the high concentration of BSA.
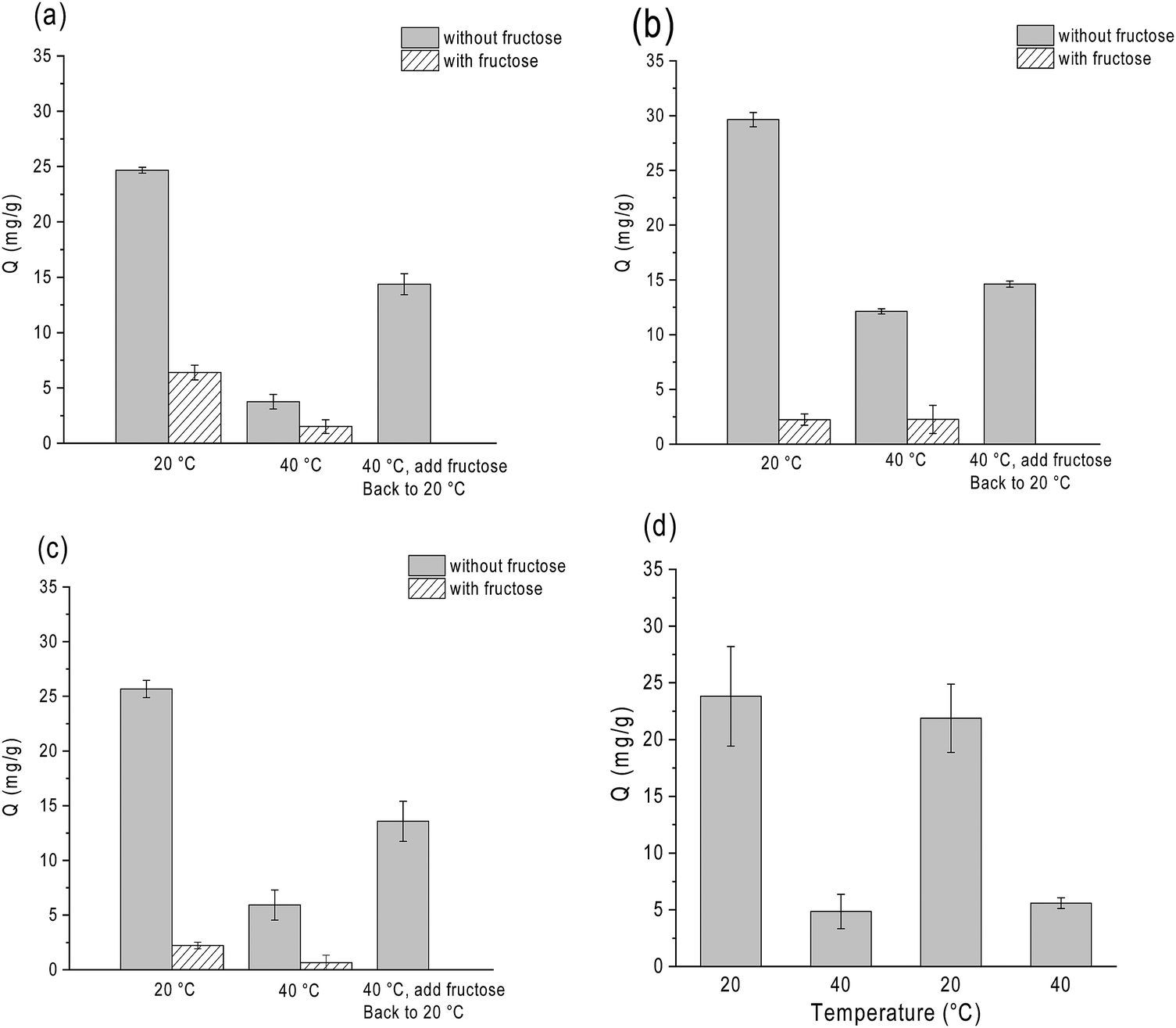 | ||
| Fig. 6 Controlling HRP binding by temperature and fructose. The boronic acid-functionalized particles Si@pco-BA@BA (a), Si@pco-OH@BA (b) and Si@pNIPAm@BA (c) were mixed with the protein solution at 20 °C in pH 9 buffer to measure the HRP binding. After this step the samples were heated to 40 °C before the HRP binding was measured. For the last bar, 1 mg of fructose was added at 40 °C to the initially fructose-free mixture before the sample was cooled to 20 °C. In (d) the HRP binding to Si@pNIPAm@BA was measured from a sample undergoing repeated alteration of temperature between 20 and 40 °C. | ||
Using Si@pNIPAm@BA as an example, we investigated the reversibility of the temperature-controlled HRP binding to boronic acid-functionalized polymer brushes. Fig. 6d shows that after the solution temperature was increased from 20 °C to 40 °C, HRP binding decreased from 23 mg g−1 to 4 mg g−1. After the solution temperature returned to 20 °C, in the same sample the HRP binding went back to the initial value. This result indicates clearly that the temperature-controlled glycoprotein binding and release are reversible. We should note that for the nanoparticle-supported polymer brushes that did not contain the thermo-responsive NIPAm component (Si@pGMA-BA@BA and Si@pGMA-OH@BA), variation of temperature did not affect the HRP binding (Fig. S5, ESI†). The possibility of controlling protein binding via temperature is interesting for future exploration, for example to study selective loading and release of protein pharmaceuticals in thermo-responsive polymer brushes.
Boronate affinity chromatography is a proven technique for purification of glycoproteins.23–28 In general terms, separation of glycoproteins through reversible boronate ester bonds does not cause protein denaturation. In the protein binding experiment, we quantified the glycoprotein HRP by measuring its enzyme activity. The temperature-recycling experiment (Fig. 6d) demonstrated that the HRP binding and releasing could be controlled by temperature variation, and the binding-releasing event was truly reversible. As the enzyme activity of the released protein resumed after returning to the original temperature, it is very clear that the protein function before and after the binding was not affected.
Controlling the release of glycoprotein from boronate affinity particles
As mentioned already in previous sections, variation of pH is often used to control glycoprotein binding to boronate affinity materials. To investigate how several effectors can be combined to influence glycoprotein release, we studied the binding and release of HRP on Si@pco-BA@BA in the presence of excess of BSA under different solvent conditions. The composite particles were first loaded with HRP in the binary protein solution in pH 9 buffer and washed to remove the non-specifically adsorbed protein. The glycoprotein was then eluted from the particles using pH 4 buffer at different temperatures, with and without the addition of fructose in the elution buffer. As shown in Fig. 7, although elution with pH 4 buffer could release HRP, adding fructose in the elution buffer increased the protein recovery by almost 50%. When the protein elution with pH 4 buffer was carried out at 40 °C, we obtained the highest HRP recovery irrespective of fructose. This result points to a more efficient means of glycoprotein separation by combining acidic buffer with thermo-desorption, which can be easily applied to thermo-responsive boronate affinity materials.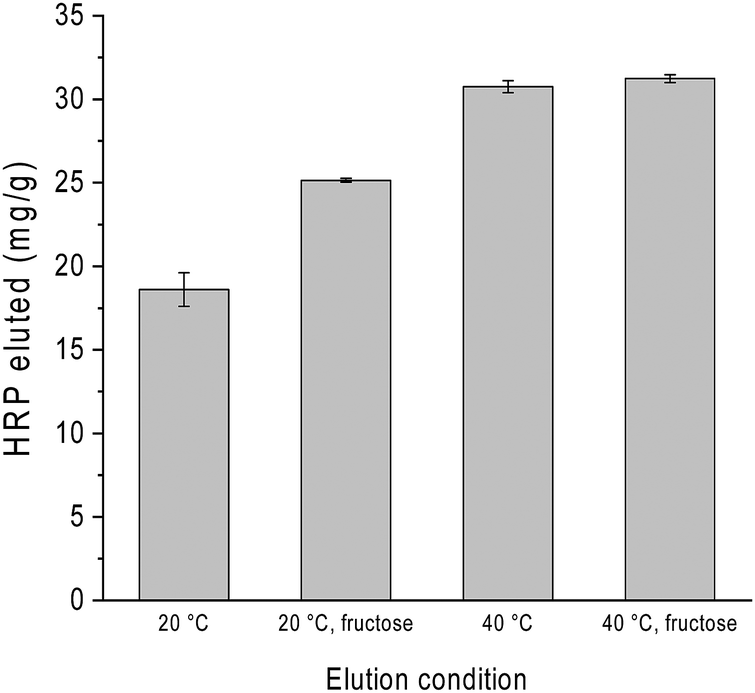 | ||
| Fig. 7 Elution of HRP from Si@pco-BA@BA using acidic buffer under different conditions. | ||
In this work the boronic acids were immobilized on the polymer brushes through stable covalent bonds. Consequently, no boronic acid will detach from the composite particles to associate with the protein in the elution step. As the binding of glycoprotein to the boronic acid-modified polymer brushes was through a carbohydrate moiety, the glycoproteins are expected to maintain their structure, as we have demonstrated in a previous work where a bound glycoenzyme (HRP) maintained its catalytic activity.42
Conclusions
In this work, we synthesized five different types of boronic acid-functionalized polymer brushes on silica nanoparticles to study their structure–function relationship. We also investigated the use of temperature variation to control the binding and release of glycoproteins from boronate affinity particles. For polymer brushes designed to bind glycoproteins, our results suggest that the most important boronic acids contributing to protein binding are located at the chain termini of the polymer brushes. For the thermo-responsive polymer brushes, we demonstrated that it is possible to use temperature to control the glycoprotein binding and release. The reversible characteristic of the boronate ester bonds involved in the protein separation process has been revealed through temperature recycling experiments. For the first time, we have shown that glycoproteins bound on boronate affinity particles can be eluted by simply varying the solution temperature. When combined with acidic buffer, the new thermo-desorption provided the highest protein recovery. The new boronic acid-functionalized, nanoparticle-supported polymer brushes hold interesting potential to be exploited not only for protein separation, but also for other applications including controlled drug delivery and release.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Foundation for International Cooperation in Research and High Education (STINT, grant no. CH2015-6254), the Swedish Research Council FORMAS (grant no. 2016-00591) and the National Natural Science Foundation of China (NSFC, grant no. 21611130030). Huiting Ma thanks the China Scholarship Council (201606160072) for a visiting scholarship. Huiting Ma is a visiting student in Lund University and a PhD candidate in the Huazhong University of Science and Technology.References
- S. G. Boyes, B. Akgun, W. J. Brittain and M. D. Foster, Macromolecules, 2003, 36, 9539–9548 CrossRef.
- X. Kong, T. Kawai, J. Abe and T. Iyoda, Macromolecules, 2001, 34, 1837–1844 CrossRef.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef.
- Y.-P. Wang, X.-W. Pei and K. Yuan, Mater. Lett., 2005, 59, 520–523 CrossRef.
- T. von Werne and T. E. Patten, J. Am. Chem. Soc., 1999, 121, 7409–7410 CrossRef.
- L. Zhang and M. Fang, Nano Today, 2010, 5, 128–142 CrossRef.
- F. Zhou and W. T. Huck, Phys. Chem. Chem. Phys., 2006, 8, 3815–3823 RSC.
- C. Perruchot, M. Khan, A. Kamitsi, S. V. Armes, T. von Werne and T. Patten, Langmuir, 2001, 17, 4479–4481 CrossRef.
- Z. Zhou, S. Zhu and D. Zhang, J. Mater. Chem., 2007, 17, 2428–2433 RSC.
- S. Zhang, W. Guo, J. Wei, C. Li, X.-J. Liang and M. Yin, ACS Nano, 2017, 11, 3797–3805 CrossRef PubMed.
- I. Y. Galaev and B. Mattiasson, Trends Biotechnol., 1999, 17, 335–340 CrossRef PubMed.
- G. Pasparakis and M. Vamvakaki, Polym. Chem., 2011, 2, 1234–1248 RSC.
- L. Jiang, M. E. Messing and L. Ye, ACS Appl. Mater. Interfaces, 2017, 9, 8985–8995 Search PubMed.
- J. Zhang, T. He, L. Tang and Z. Q. Zhang, J. Sep. Sci., 2016, 39, 1691–1699 CrossRef PubMed.
- L. Zhang, Y. Xu, H. Yao, L. Xie, J. Yao, H. Lu and P. Yang, Chem. – Eur. J., 2009, 15, 10158–10166 CrossRef PubMed.
- E. Robertson and J. Kennedy, Bioseparation, 1996, 6, 1–15 Search PubMed.
- F. M. Okanda and Z. El Rassi, Electrophoresis, 2006, 27, 1020–1030 CrossRef PubMed.
- M. Madera, Y. Mechref and M. V. Novotny, Anal. Chem., 2005, 77, 4081–4090 CrossRef PubMed.
- M. Bedair and Z. El Rassi, J. Chromatogr. A, 2004, 1044, 177–186 CrossRef PubMed.
- P. M. Drake, B. Schilling, R. K. Niles, M. Braten, E. Johansen, H. Liu, M. Lerch, D. J. Sorensen, B. Li and S. Allen, Anal. Biochem., 2011, 408, 71–85 CrossRef PubMed.
- X. Mao, Y. Luo, Z. Dai, K. Wang, Y. Du and B. Lin, Anal. Chem., 2004, 76, 6941–6947 CrossRef PubMed.
- C. Zhang, C. Bi, W. Clarke and D. S. Hage, J. Chromatogr. A, 2017, 1523, 114–122 CrossRef PubMed.
- Y. Qu, J. Liu, K. Yang, Z. Liang, L. Zhang and Y. Zhang, Chem. – Eur. J., 2012, 18, 9056–9062 CrossRef PubMed.
- K. Sparbier, S. Koch, I. Kessler, T. Wenzel and M. Kostrzewa, J. Biomol. Tech., 2005, 16, 407–413 Search PubMed.
- Y. Liu, L. Ren and Z. Liu, Chem. Commun., 2011, 47, 5067–5069 RSC.
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 42, 1106–1123 RSC.
- D. Li, Y. Chen and Z. Liu, Chem. Soc. Rev., 2015, 44, 8097–8123 RSC.
- Z. Liu and H. He, Acc. Chem. Res., 2017, 50, 2185–2193 CrossRef PubMed.
- L. Ren, Z. Liu, M. Dong, M. Ye and H. Zou, J. Chromatogr. A, 2009, 1216, 4768–4774 CrossRef PubMed.
- H. Wang, Z. Bie, C. Lü and Z. Liu, Chem. Sci., 2013, 4, 4298–4303 RSC.
- X. Zhang, X. He, L. Chen and Y. Zhang, J. Mater. Chem. B, 2014, 2, 3254–3262 RSC.
- W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572–2590 CrossRef.
- Y. Xu, Z. Wu, L. Zhang, H. Lu, P. Yang, P. A. Webley and D. Zhao, Anal. Chem., 2008, 81, 503–508 CrossRef PubMed.
- B. J. Gould and P. M. Hall, Clin. Chim. Acta, 1987, 163, 225–230 CrossRef.
- Q. Zhang, A. A. Schepmoes, J. W. Brock, S. Wu, R. J. Moore, S. O. Purvine, J. W. Baynes, R. D. Smith and T. O. Metz, Anal. Chem., 2008, 80, 9822–9829 CrossRef PubMed.
- T. Suksrichavalit, K. Yoshimatsu, V. Prachayasittikul, L. Bülow and L. Ye, J. Chromatogr. A, 2010, 1217, 3635–3641 CrossRef PubMed.
- L. Jiang, H. Bagán, T. Kamra, T. Zhou and L. Ye, J. Mater. Chem. B, 2016, 4, 3247–3256 RSC.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef PubMed.
- H. Tu, C. E. Heitzman and P. V. Braun, Langmuir, 2004, 20, 8313–8320 CrossRef PubMed.
- T. Kawai, K. Saito and W. Lee, J. Chromatogr. B: Biomed. Sci. Appl., 2003, 790, 131–142 CrossRef.
- K. Ohno, T. Morinaga, S. Takeno, Y. Tsujii and T. Fukuda, Macromolecules, 2007, 40, 9143–9150 CrossRef.
- H. Bagán, T. Kamra, L. Jiang and L. Ye, Macromol. Chem. Phys., 2017, 218, 1600432 CrossRef.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- J. A. Huntington and P. E. Stein, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 756, 189–198 CrossRef.
- N. C. Veitch, Phytochemistry, 2004, 65, 249–259 CrossRef PubMed.
- K. A. Majorek, P. J. Porebski, A. Dayal, M. D. Zimmerman, K. Jablonska, A. J. Stewart, M. Chruszcz and W. Minor, Mol. Immunol., 2012, 52, 174–182 CrossRef PubMed.
- L. Li, Y. Lu, Z. Bie, H. Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2013, 52, 7451–7454 CrossRef PubMed.
- W. Wang, X. Gao and B. Wang, Curr. Org. Chem., 2002, 6, 1285–1317 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb00627j |
| This journal is © The Royal Society of Chemistry 2018 |