
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombinatorial delivery of bioactive molecules by a nanoparticle-decorated and functionalized biodegradable scaffold†
Ewa M.
Czekanska‡
a,
Jin
Geng‡
 b,
Michael
Glinka
a,
Kate
White
a,
Janos
Kanczler
a,
Nicholas D
Evans
a,
Richard O. C.
Oreffo
*a and
Mark
Bradley
*b
b,
Michael
Glinka
a,
Kate
White
a,
Janos
Kanczler
a,
Nicholas D
Evans
a,
Richard O. C.
Oreffo
*a and
Mark
Bradley
*b
aBone and Joint Research Group, Centre for Human Development, Stem Cells and Regeneration, Faculty of Medicine, Southampton University, Southampton, SO16 6YD, UK. E-mail: Richard.Oreffo@soton.ac.uk
bSchool of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: mark.bradley@ed.ac.uk
First published on 14th June 2018
Abstract
The combination of supportive biomaterials and bioactive factors to stimulate endogenous progenitor cells is of key interest for the treatment of conditions in which intrinsic bone healing capacities are compromised. To address this need a “scaffold-decoration platform” was developed in which a biocompatible, biotin-functionalised 3D structural polymer network was generated through a solvent blending process, and used to recruit avidin modified nanoparticles within its 3D structure through biotin–avidin conjugation. This was enabled via the generation of a suite of poly(lactic-co-glycolic acid) (PLGA) nanoparticles, encapsulating two bioactive factors, vascular endothelial growth factor (VEGF) and L-ascorbic acid 2-phosphate (AA2P) and conjugated to streptavidin to allow attachment to the bone generating scaffold. The levels of encapsulated and released VEGF and AA2P were tailored to fall within the desired range to promote biological activity as confirmed by an increase in endothelial cell tubule formation and collagen production by osteoblast cells in response to nanoparticle release of VEGF and AA2P, respectively. The release of VEGF from the scaffolds produced a significant effect on vasculature development within the chick chorioallantoic membrane (CAM) angiogenic assay. Similarly, the scaffolds showed strong biological effects in ex vivo assays indicating the potential of this platform for localised delivery of bioactive molecules with applications in both hard and soft tissue engineering.
Introduction
The current paradigm for tissue engineering consists of 3 elements, (i) cells, (ii) an appropriate scaffold and, (iii) conditioning of the cells and scaffolds with appropriate mechanical forces and factors to create a tissue equivalent in an in vivo like environment.1,2Following severe bone trauma or bone loss as a consequence of ageing or pathological conditions, the application of tissue engineering and regenerative medicine has come to the fore with the aspiration to aid bone regeneration and health and, ultimately, to restore the anatomical function and structure of bone. A central requirement in such situations is the ability to support the function of the cells present in bone to produce extracellular matrix essential for mineral deposition as well as to support the development of blood vessels.3 It has long been known that the processes of angiogenesis and osteogenesis in bone are closely interrelated and pivotal in bone development, growth and regeneration.4 The current principles of bone regeneration places vasculature formation as a critical factor for successful bone healing with the lack of vasculature the main cause of delayed union or non-union.5,6 Vasculature endothelial growth factor (VEGF) is essential for vasculature formation by endothelial cells and influences the proliferation and differentiation of bone-forming cells.7–10 Furthermore, the function of bone cells is exquisitely regulated through interaction with the collagenous extracellular matrix (ECM) including interactions with the central bone organic component, collagen, the formation of which, in turn, is regulated by ascorbic acid.11,12
The copolymer poly(lactic-co-glycolic acid) (PLGA) has received considerable attention as a delivery system given its excellent biocompatibility, high safety profile, and the Federal Drug Administration approval of PLGA for use in drug delivery.13–16 In addition, PLGA is typically degraded into oligomers and monomers, which are natural metabolites.17 Previous studies have shown the efficacious nature of PLGA to allow the delivery of proteins in comparison to free protein.18,19
The encapsulation of bioactive substances within particles at the nanometre scale confers several advantages over microparticle encapsulation, including a reduced risk of embolisation and enhanced rates of release due to large surface area to volume ratios.20,21 VEGF delivery via PLGA nanoparticles (PLGA NP) has already shown promise in encouraging blood vessel growth in tissue engineering and cardiovascular medicine applications.22 It however important to ensure that the delivered biologically active molecules are released at the site of tissue repair for extended periods of time, while retaining biological activity over this timeframe. Strategies such as integrating nanoparticle into polymeric hydrogels23,24 and bioactive glass scaffolds25,26 have been developed to support controlled delivery of bioactive molecules. A co-delivery system for VEGF and basic FGF, loaded PLGA nanoparticles incorporated within a hydrogel has been reported by Jiang and colleagues to enhance tissue regeneration.27 Li and co-workers have shown the delivery of BSA-encapsulated BMP2 and dexamethasone within a polymeric nanofiber scaffold in vivo resulting in increased new bone formation compared to blank scaffolds.28 We hypothesised that scaffolds decorated with nanoparticles presented on the scaffold surface would provide an attractive and accessible delivery platform for cells with a stable delivery system and high local concentrations being supported by nanoparticle retention.
In our previous work, ternary mixtures of natural and synthetic polymer blends displayed promising potential as a bone tissue engineering matrix for bone regeneration.29 In this study, PLGA-based nanoparticles containing VEGF and ascorbate were individually prepared and evaluated to determine their efficiency of release in vitro. Subsequently the nanoparticles were combined with a biotin-tagged polymer-blend scaffold to allow linkage of the avidin-loaded nanoparticles onto the biotin-conjugated scaffold thus allowing stable and controlled release of two bioactive molecules. This was examined in vivo using the chick chorioallantoic angiogenic membrane assay.
Results and discussion
Preparation of polymer scaffolds functionalised with nanoparticles
The current studies demonstrate that the combination of nanoparticles containing bioactive molecules within biotin-tagged polymer-blend scaffolds provides a platform for the localised, and spatial, delivery of bioactive molecules with applications in both hard and soft tissue engineering.NHS–palmitic acid was conjugated to free amino groups on avidin (0.1 equivalent to palmitic acid) producing a stable amide linkage and rendering the protein hydrophobic while the presence of 2% (w/v) deoxycholate prevented palmitate vesicle formation (Fig. 1A). Biotinylated polymers were generated by treating biotin with triethylamine and N,N′-disuccinimidyl carbonate at room temperature for 16 hours, followed by the addition of polyethyleneimine (PEI) to the mixture and subsequent PEI–biotin isolation (Fig. 1B). Based on the 1H NMR of PEI–biotin (see Fig. S1, ESI†), around 14 biotin units were attached per PEI unit. PLGA NPs loaded with VEGF and AA2P were prepared using a double emulsion method in the presence of avidin–palmitate allowing the hydrophobic chain to embed into the PLGA nanoparticles. Particle size analysis confirmed their submicron size and indicated a relatively narrow particle size distribution (660 ± 22 nm) with only minor changes in particle size when loaded with different cargos. The PLGA nanoparticles displayed zeta-potentials ranging from −20.3 mV to −32.1 mV, which can be attributed to the presence of carboxylate groups on the surface of the nanoparticle, due to the use of acid terminated PLGA polymers (Table 1).
 | ||
| Fig. 1 Synthesis of (A) an avidin–palmitic acid conjugate and. (B) Biotinylated-PEI. | ||
| PLGA | Cargo | Mean diameter (nm) | PDi | Zeta potential (mV) |
|---|---|---|---|---|
| 38 kDa | RhB | 672 | 0.09 | −25.2 |
| 38 kDa | — | 675 | 0.10 | −20.3 |
| 50 kDa | RhB | 637 | 0.12 | −20.8 |
| 7 kDa | RhB | 653 | 0.15 | −32.1 |
| 38 kDa | VEGF | 682 | 0.10 | −29.1 |
| 38 kDa | AA2P | 669 | 0.11 | −28.2 |
A bone repair polymer blend was generated by encompassing a number of biodegradable polymers, chitosan (CS), poly-L-lactide (PLLA) and polyvinyl acetate (PVAc) (Fig. 2A), based on our previous reported work.29 To generate biotin functionalised polymer scaffolds while retaining the cellular binding affinity, a low percentage of PEI–biotin was used in the polymer blends (PLLA/PVAc/CS/PEI–biotin, 25/25/10/2, w/w). The biotin modified polymer scaffolds were fabricated by solvent blending PEI–biotin with CS, PLLA and PVAc followed by freeze-drying (see Table 2 for ratios of the polymers), with solid–liquid phase separation creating a porous scaffold upon solvent removal. The scaffolds were loaded with nanoparticles by incubation of the avidin-modified PLGA NPs with the biotinylated-polymer scaffold in PBS at room temperature (Fig. 2B). The morphology of the nanoparticles loaded with rhodamine B (RhB) as analysed by scanning electron microscopy (SEM) revealed a uniform spherical structure (Fig. 3A). Crucially, the NP structures remained intact following incorporation within the polymer scaffolds (Fig. 3B), with cross-sectional SEM analysis demonstrating an even distribution of intact PLGA-NPs within the polymer network (Fig. 3C). The polymer blend scaffolds displayed a macro-porous framework, with an overall porosity of 57% ± 3.21% and 61% ± 4.53%, before and after nanoparticle decoration, respectively.
 | ||
| Fig. 2 (A) Scaffold preparation. The four polymers; polyvinyl acetate, chitosan, poly(L-lactide) and PEI–biotin were solvent blended as previously described.29 (B) Scaffold decorated combinatorially with nanoparticles. The image shows the polymer scaffold (17 mm × 2 mm) combined with PLGA nanoparticles. | ||
| Polymer | Concentration (w/v%) | Volume (ml) |
|---|---|---|
| PVAc | 10% in chloroform | 2.5 |
| PLLA | 10% in chloroform | 2.5 |
| CS | 1% in water with 2% acetic acid | 10 |
| PEI–biotin | 20% in water | 0.1 |
 | ||
| Fig. 3 Morphology, size and distribution of NPs in the PLGA scaffolds. SEM images of (A) avidin labelled nanoparticles, (B) biotin labelled scaffold and (C) scaffolds combined with nanoparticles. NPs were uniform and had narrow size distribution. Nanoparticle structures remained intact after incorporation within the polymer scaffolds. Scale bars represent 1 μm for image (A) and (B); 4 μm for image (C). | ||
Release profiles of AA2P and VEGF
A range of PLGA compositions with different molecular weights and monomer compositions (7 kDa 50/50, 38 kDa 50/50, and 50 kDa 85/15) were employed to study biofactor release profiles. In the first instance, the release kinetics of rhodamine B (RhB) encapsulated within NPs generated from the three PLGA polymers were evaluated in vitro (Fig. 4A).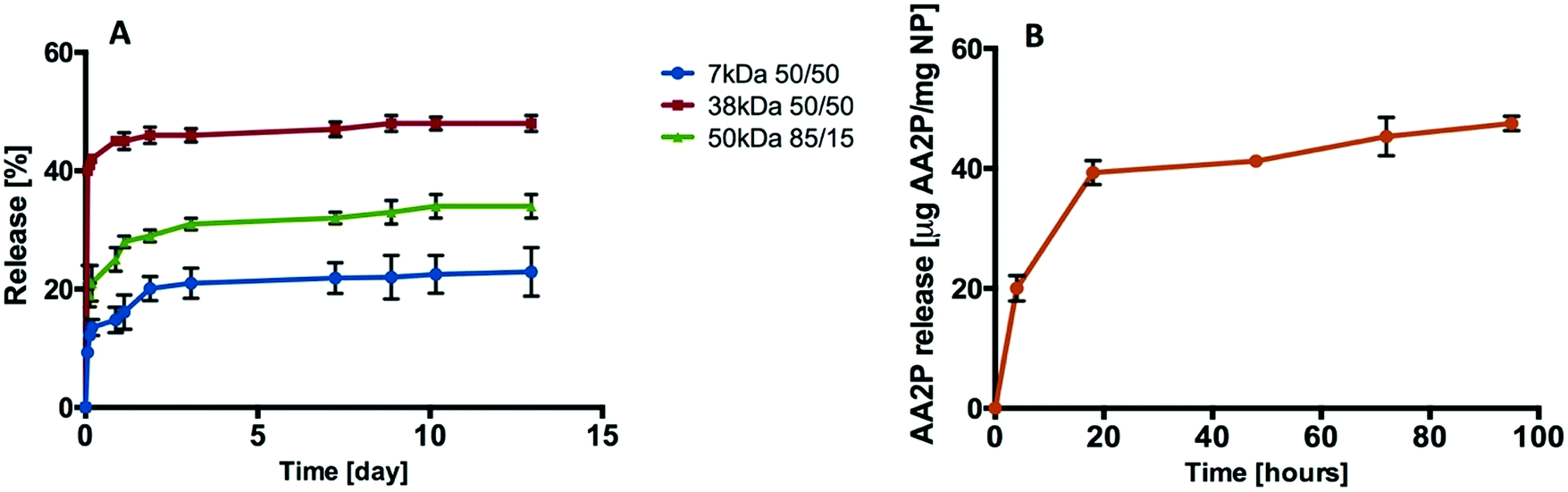 | ||
| Fig. 4 Release profiles. (A) Release of rhodamine B from PLGA (7 kDa, 38 kDa, and 50 kDa) nanoparticles. (B) Release of AA2P from PLGA (38 kDa 50/50) nanoparticles (n = 3; one batch). | ||
The release of RhB from PLGA-NP reached 10%, 40% and 22% from the 7 kDa, 38 kDa and 50 kDa polymers respectively during the first 1.5 hours. After 12 days, 48% of the RhB was observed to have been released from the 38 kDa PLGA-NP compared to 22% from the 7 kDa and 34% from the 50 kDa PLGA-NPs (Fig. 4A). Based on this data, 38 kDa PLGA was selected to encapsulate VEGF and L-ascorbic acid 2-phosphate (AA2P) in all subsequent studies.
Loading of AA2P or VEGF into the PLGA nanoparticles was achieved using a double emulsion method. Burst release was observed for AA2P within 20 hours followed by gradual release of AA2P over 96 hours (Fig. 4B). In initial experiments, the release of VEGF from PLGA nanoparticles was monitored over 10 days (Fig. 5A). Initial burst release was reported over the first 30 minutes, but over a 72 hour time frame, VEGF release was observed to steadily increase, reaching a plateau with a concentration of 81 ng VEGF per mg of NPs (Fig. 5B).
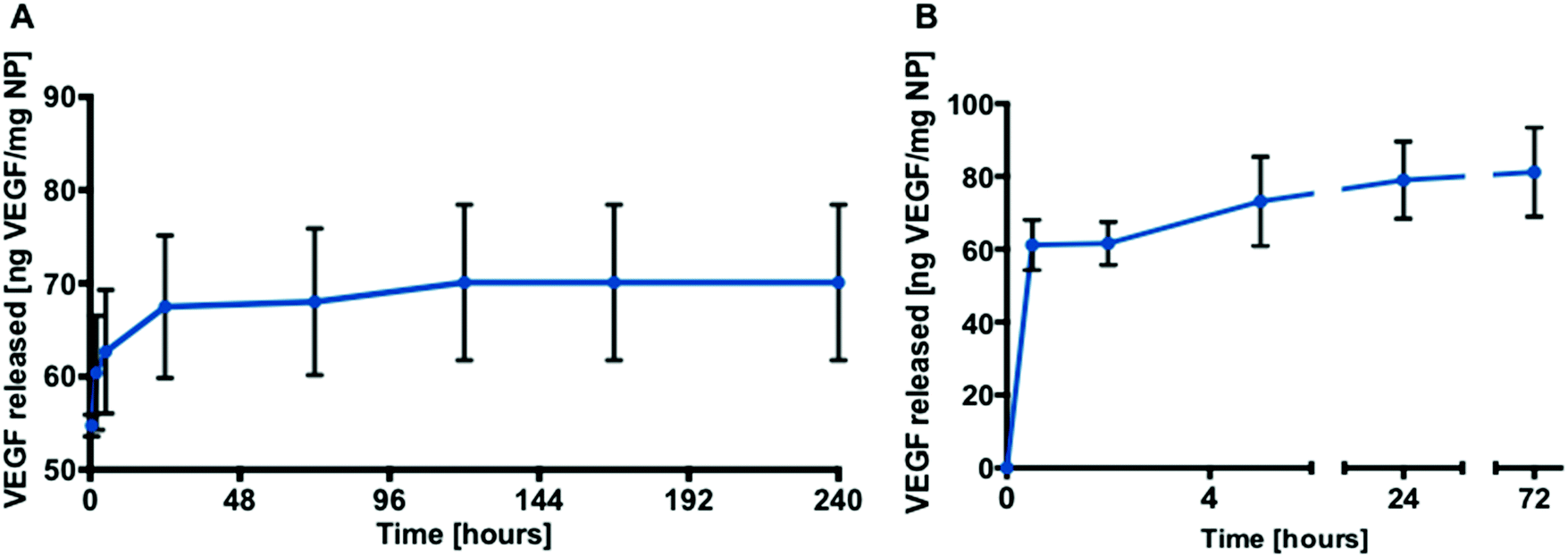 | ||
| Fig. 5 Release profiles. (A) Release of VEGF from PLGA (38 kDa 50/50) nanoparticles over 10 days (n = 3; one batch). (B) Release of VEGF from PLGA (38 kDa 50/50) nanoparticles over 72 hours (n = 9; three separate studies). The data are presented as mean ± SD. | ||
The current studies also assessed the release profiles of AA2P from scaffolds containing nanoparticles functionalised with and without avidin. We observed the release of 28 μg and 14 μg of AA2P released from one scaffold containing nanoparticles decorated with and without avidin, respectively (Fig. S5, ESI†). Since the avidin functionalised nanoparticles will be attached to the polymer scaffolds, as a consequence of avidin–biotin binding, scaffolds decorated with avidin functionalised nanoparticles would be expected to show increased collagen release as observed (Fig. S6, ESI†).
Bioactivity analysis of AA2P and VEGF released from nanoparticles
Ascorbic acid (vitamin C) is a co-factor for lysyl and prolyl hydroxylase, two essential enzymes in the collagen biosynthesis pathway.11 Similar attributes are associated with its derivatives, including 2-phosphate-L-ascorbic acid (AA2P) which exhibits higher stability in solution.30 Limited collagen biosynthesis, due to ascorbic acid deficiency, manifests in various systemic pathologies, including abnormalities in bone maturation, epiphyseal disease, pseudoparalysis related to bone pain, spontaneous fracturing and impaired bone healing.31 Therefore, the delivery of ascorbic acid or AA2P in the fracture environment could stimulate the healing process. Collagen deposition by MC3T3-E1 cells was observed to increase after 7 days culture, following supplementation with 2-phosphate-L-ascorbic acid or nanoparticles containing 2-phosphate-L-ascorbic acid (Fig. 6). There was a significant 7.24 (p < 0.01) and 5.8-fold (p < 0.05) increase in collagen production in MC3T3-E1 cultures maintained in medium containing 0.5 mg NP-AA2P compared to blank NPs and the no treatment control respectively. In addition, a 6.95-fold (p < 0.05) increase in collagen production compared to the NP-blank was observed in cell cultures supplemented with 1 mg NP-AA2P (Fig. 6). It is important to note that the concentration of 1 μg AA2P per ml added to medium was reported to be sufficient to increase collagen production in MC3T3-E1 cells in a long-term manner and further addition of AA2P was not observed to correlate with collagen production.32 Calculations based on the release kinetics of NP-AA2P showed that the concentration of released AA2P from 0.5 mg NP-AA2P was 15 μg ml−1 (Fig. 4B). Initial studies focusing on the effect of ascorbic acid and its derivatives on collagen production in vitro reported dose–response effects within 3 days that correspond to short-term effects, while long-term effects were not evaluated.33,34 Although, the administration of higher AA2P concentrations could have a beneficial effect on the earlier onset of cell differentiation compared to lower concentrations, in the long term, concentrations of 1 μg AA2P per ml would appear sufficient to promote collagen production. Hence, the delivery of NP-AA2P could have promising effects on both, stimulation of collagen production and cell differentiation in vivo.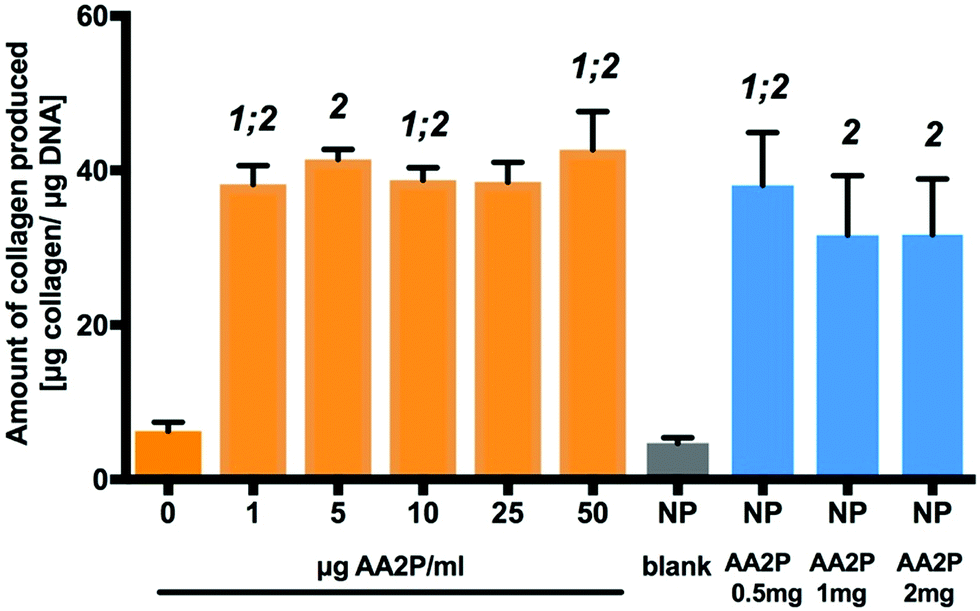 | ||
| Fig. 6 Bioactivity of released AA2P from PLGA-NPs. Fold-changes in collagen production in response to AA2P supplemented into medium and released from the nanoparticles. 1: statistically different from 0 μg AA2P per ml; 2: statistically significant from NP blank. Statistical significance at p < 0.05 (n = 9, three separate studies). The data are presented as mean ± SD. | ||
Tubule branch length formed by HUVECs was found to correlate with VEGF concentrations added to the medium. A two-fold increase (±0.6; p < 0.0001) in HUVEC tubule branch length was observed at 40 ng VEGF per ml, compared to results obtained from cultures in the absence of VEGF (Fig. 7). The increase in HUVEC tubule branch length was 2 (p < 0.001) and 3-fold (p < 0.0001) for cells maintained in medium containing 0.5 mg NP-VEGF compared to cultures in the absence of VEGF or the NP-blank, respectively. There was no significant difference in the branching length for groups maintained in medium containing 1 mg NP-VEGF compared to control (0 ng VEGF per ml) and conditioned medium (CM) control groups. The concentration of VEGF released into medium form the NPs was approximately 70 ng VEGF per ml for the CM 1 group and 35 ng VEGF per ml for the CM 0.5 group as evidenced by ELISA (Fig. 5B). The lower concentration of VEGF was sufficient to stimulate endothelial tubular network formation and to support physiological vessel growth and limit vessel leakage.35,36 Controlling the dosage of VEGF released from NPs on a micro-environmental level would support optimal tubular network formation and angiogenesis in vivo.
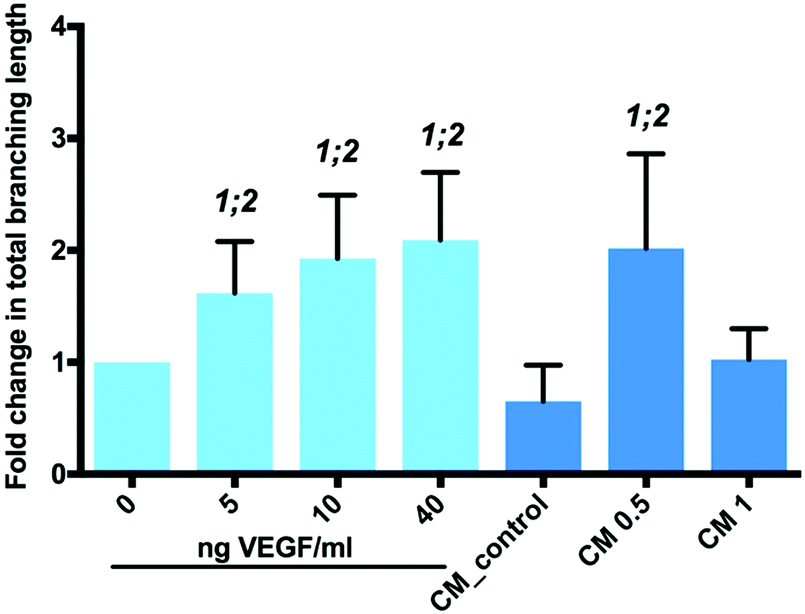 | ||
| Fig. 7 Bioactivity of VEGF released from PGLA-NP's. Fold changes in total branching length of tubules formed in 2D HUVEC tubule formation assay on 3% LAPONITE®-fibronectin hydrogel. CM: conditioned medium; CM 0.5: conditioned medium after incubation with 0.5 mg NP-VEGF for 6 hours; CM 1: conditioned medium after incubation with 1 mg NP-VEGF for 6 hours. 1: statistically different from 0 ng VEGF per ml; 2: statistically significant from CM control. Statistical significance at p < 0.05 (n = 9, three separate studies). The data are presented as mean ± SD. | ||
Chorioallantoic membrane (CAM) assay of polymer-nanoparticle scaffolds for angiogenic bioactivity
The chorioallantoic membrane (CAM) assay was used to determine the potential of VEGF-NPs to enhance angiogenesis after 8 days culture. Scaffolds were found to be fully integrated within the CAM and vessel formation was clearly visible (Fig. S8, ESI†).The activity of released VEGF from the nanoparticles contained within the scaffold was evaluated using the CAM assay (Fig. 8A). The Chalkley score method was used to evaluate changes in the vasculature of the CAM in the presence of VEGF. An increase in vessel number was observed in CAM retrieved from eggs treated with 25 and 100 ng VEGF and CAM samples with scaffolds containing VEGF-NPs (Fig. 8A). The addition of VEGF to CAM at embryonic day 10, resulted in a significant dose-dependent increase in the Chalkley score at day 18 compared to the control; generating a Chalkley score of 5.1 (±0.8) for controls and for 25 and 100 ng VEGF a score of 7.8 (±1.6; p < 0.001) and 10.2 (±1.7; p < 0.001), respectively (Fig. 8A). In addition, the Chalkley score was significantly higher for the VEGF-NPs samples compared to scaffolds containing blank NPs (p < 0.001) and the no treatment group (p < 0.001). The Chalkley score was noted to be comparable for scaffolds containing VEGF-NPs (10.1 ± 2.11) compared to exogenous 10 ng VEGF (10.2 ± 1.7) (Fig. 8A).
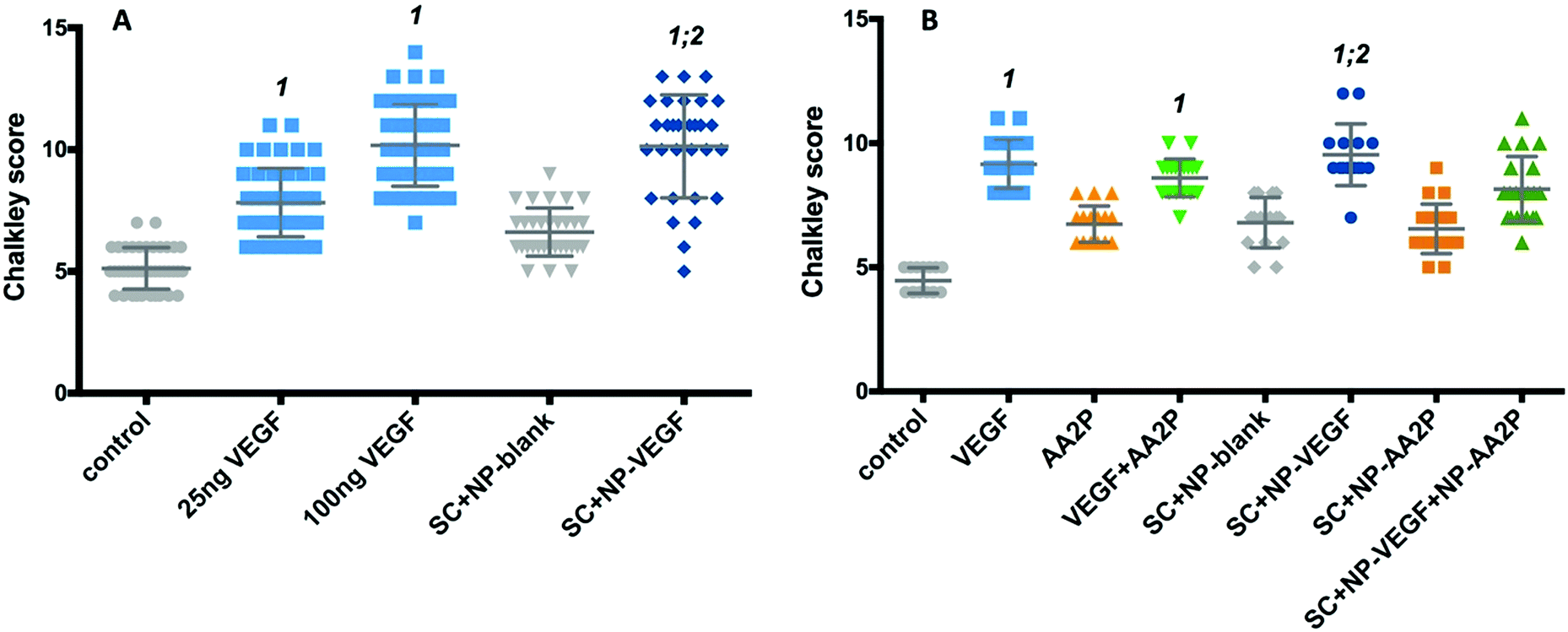 | ||
| Fig. 8 Evaluation of vasculature at day 18 of embryonic chick development using Chalkley score method in response to single- or dual-release of bioactive molecules from scaffold. (A) Chalkley point-overlap morphometry results indicating changes in vessel numbers in CAM at day 18 after 8 days incubation with VEGF, or scaffold containing NP-VEGF; (B) Chalkley point-overlap morphometry results indicating changes in vessel numbers in CAM at day 18 after 8 days incubation with VEGF, AA2P or scaffolds containing NP-VEGF and/or AA2P. No VEGF and scaffolds with blank NP were used as controls. SC + NP-blank – scaffold containing PLGA nanoparticles; SC + NP-VEGF – scaffold containing nanoparticles with VEGF; SC + NP-AA2P – scaffold containing nanoparticles with AA2P; SC + NP-VEGF + NP-AA – scaffold containing nanoparticles with VEGF and AA2P; control – no treatment; VEGF – CAM treated with 25 ng VEGF; AA2P – CAM treated with 50 μg VEGF. 1: statistically different from control; 2: statistically significant from SC + NP-blank. Statistical significance at p < 0.05 (9–12 samples per group; three separate studies). The data are presented as mean ± SD. | ||
Finally, we examined angiogenesis in CAM cultures treated with scaffolds containing nanoparticles encapsulated with VEGF and nanoparticles with AA2P. A significant increase in the Chalkley score observed for VEGF-NPs, compared to control (CAM only; p < 0.0001) and scaffold containing blank-NPs (p < 0.001), was observed (Fig. 8B). The release of AA2P alone did not affect angiogenesis, while the combined release of AA2P and VEGF indicated an increased vasculature network evidenced by the increase in Chalkley score compared to CAM control and scaffolds containing blank-NPs or AA2P-NPs (Fig. 8B), although, this did not reach statistical significance.
Discussion
The current major challenge in using synthetic scaffold materials for tissue engineering lies in the requirement for growth factor delivery mechanisms that mimic the in vivo release profiles of factors produced during natural tissue morphogenesis or repair. The biocompatibility of PLGA based-microparticles and scaffold delivery systems were shown via the bioactivity of the encapsulated growth factor VEGF and the long-lasting analogue of ascorbic acid, 2-phosphate-L-ascorbic acid (AA2P).22,37,38 The delivery of multiple growth factors from soft and hard polymer scaffolds with distinct kinetics has been reported previously.39 However, the administration of multiple growth factors from polymer scaffolds via the incorporation of PLGA nanoparticles allows for more precise delivery and control of release that has not been shown before. In the current study, we used a combination of surface modified PLGA nanoparticles and biotin functionalised polymer scaffolds. Palmitic acid was coupled to avidin, which then was introduced onto the surface of PLGA nanoparticles. Biotinylated PEI, PVAc, PLLA and chitosan were solvent blended into 3D structural polymer networks. VEGF and AA2P were encapsulated within the PLGA nanoparticles, which were loaded onto the polymer network/scaffolds through biotin–avidin conjugation. The release profiles of PLGA nanoparticles was controlled by using PLGA polymers with different molecular weights and monomer compositions. The in vitro release profiles from PLGA nanoparticles showed a moderate initial burst release followed by a sustained release. The release of VEGF and AA2P from the nanoparticles alone, and when attached to the scaffolds, confirmed the bioactivity of both factors/molecules within in vitro and ex vivo/in vivo environments. The limitation of using PLGA nanoparticle as a delivery system has been discussed in previous studies.39 A more controlled release system, such as triggering release, will potentially improve the lack of control of release by using PLGA nanoparticles that limited correlation between growth factor efficacies in the current study. Given the fundamental role of vascularisation in the bone healing process and production of extracellular matrix,11,40 the activity of released VEGF and AA2P to stimulate vascularisation and collagen production, respectively, was addressed and evaluated. The release of VEGF stimulated tubule formation by endothelial cells and vasculature formation in CAM, while AA2P stimulated collagen deposition by osteoblasts in vitro. Thus, the current studies indicate the delivery of biomolecules within nanoparticles combined with scaffolds offers a promising platform to support angiogenesis and collagenous matrix formation during the bone healing process. Interestingly, the combination of VEGF and AA2P are particularly favourable for the treatment of osteoporotic patients whose bone is characterised by declining trabecular bone.41 While VEGF could stimulate angiogenesis and thus aid nutrient-waste exchange, AA2P administration could stimulate osteoblast function and, ultimately, the formation of bone. Further evaluation of scaffolds loaded with a combinatorial mixture of NPs within an in vivo environment will allow evaluation of the potential and direct effect of the proposed strategy on bone healing.Experimental section
Materials
Dulbecco Modified Eagle Medium, alpha-MEM, penicillin/streptomycin, DPBS were obtained from Lonza. Ascorbic acid 2-phosphate and glycerol 2-phosphate disodium salt hydrate were purchased from Sigma. Fetal bovine serum and M199 medium was obtained from Gibco. ECGS/H were from Promocell. VEGF, TGFβ3 were from Peprotech. Poly(lactide-co-glycolide) (PLGA) (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 Resomer RG 502, RG 505, RG 504, and 85:15 PLGA ester terminated Mw 50000–75000) and tween 20, biotin, chitosan (CS) (from crab shells), polyethylenimine (PEI) (average Mw = 800 Da), poly(L-lactic acid) (PLLA) (viscosity ∼1.0 dL g−1) and poly(vinyl acetate) (PVAc) (average Mw = 140 kDa), palmitic acid N-hydroxysuccinimide ester (NHS–palmitic acid) and avidin were from Sigma. All other reagents were from Sigma and used without further purification. Analytical grade chloroform, glacial acetic acid, dichloromethane (DCM) were from Thermo Fisher Scientific, UK. Emulsions were generated using a Branson sonifier 250 at 40% max amplification. 1H nuclear magnetic resonance spectra were recorded on a Bruker AVA500 spectrometer (500 MHz respectively) at 298 K in deuterated solvents.
:50 Resomer RG 502, RG 505, RG 504, and 85:15 PLGA ester terminated Mw 50000–75000) and tween 20, biotin, chitosan (CS) (from crab shells), polyethylenimine (PEI) (average Mw = 800 Da), poly(L-lactic acid) (PLLA) (viscosity ∼1.0 dL g−1) and poly(vinyl acetate) (PVAc) (average Mw = 140 kDa), palmitic acid N-hydroxysuccinimide ester (NHS–palmitic acid) and avidin were from Sigma. All other reagents were from Sigma and used without further purification. Analytical grade chloroform, glacial acetic acid, dichloromethane (DCM) were from Thermo Fisher Scientific, UK. Emulsions were generated using a Branson sonifier 250 at 40% max amplification. 1H nuclear magnetic resonance spectra were recorded on a Bruker AVA500 spectrometer (500 MHz respectively) at 298 K in deuterated solvents.
Preparation of avidin–palmitic acid conjugates
Avidin (10 mg ml−1) was reacted with 10-fold mol excess of NHS–palmitic acid in PBS (1 ml) containing 2% (w/v) deoxycholate. The mixture was sonicated briefly and gently mixed at 37 °C for 12 hours. To remove excess fatty acid and hydrolyzed ester, the mixture was dialyzed (MWCO 2 kDa) against PBS containing 0.15% deoxycholate. The resultant avidin–palmitate conjugate was analysed by reverse-phase HPLC.Biotinylated polymers
Biotin (0.50 g, 2.04 mmol) and N,N′-disuccinimidyl carbonate (0.52 g, 2.02 mmol) were dissolved in 10 ml dimethylformamide. Triethylamine (0.33 ml) was then added to the resulting solution. After reacting at room temperature for 16 hours, polyethyleneimine (PEI, Mw = 800) (1.22 g, 1:1 to biotin) was added to the mixture. After 12 hours at room temperature, the product was isolated by precipitation from THF and dried under reduced pressure at 40 °C, to give a white powder.
PLGA nanoparticle fabrication
PLGA-NPs loaded with rhodamine B (RhB), or VEGF or AA2P and were fabricated by adding 0.1 ml of RhB (1 mg ml−1 in PBS), VEGF (1 mg ml−1 in PBS) or AA2P (5 mg ml−1 in PBS) to 2 ml of PLGA (38 kDa, 50/50) in dichloromethane (DCM, 25 mg ml−1), and emulsified with a Branson sonicator for 40 s on an ice bath. 4 ml of a Tween-20 solution (1% in water) was added to the primary emulsion and sonicated for 40 s on an ice bath. The resulting emulsion was added to 50 ml of a Tween-20 solution (0.2% in water) containing the avidin–palmitic acid conjugate (0.05%) and sonicated twice for 40 seconds in an ice bath. The DCM was evaporated under reduced pressure at 40 °C, forming the PLGA-NPs. The nanoparticles were collected by centrifugation at 10000 rpm at 4 °C for 10 min, washed three times to remove non-entrapped VEGF or AA2P and then freeze dried. The amount of entrapped AA2P was determined by HPLC (UV, 245 nm) and VEGF was determined by an ELISA. The encapsulation efficiency was calculated by the following equation:Providing an encapsulation efficiency of RhB 18%, VEGF 20%, and AA2P 21%.

Release profiles
PLGA-NPs (1 mg ml−1) containing RhB or AA2P were loaded into 20 kDa MWCO dialysis tubing and dialysed against 20 ml of phosphate buffered saline (PBS, pH 7.4) in the dark at 37 °C. At specified time points, 100 μl of the dialysis buffer was collected and replaced with equal volume of fresh PBS. The concentrations of RhB present in the dialysate were determined (λex 530 nm and λem 590 nm).VEGF-NPs were re-suspended in DMEM at a concentration of 2 mg VEGF-NPs per ml and incubated under rotation at 37 °C. At specified time points, samples were centrifuged for 8 min at 12000 rpm and a 30 μl sample was collected for analysis. The volume collected for analysis was replaced by fresh DMEM. Protein content in samples was determined using the human VEGF Quantikine ELISA (R&D Systems) according to the manufacturer protocol.
Polymer scaffold fabrication
Polymer blend scaffolds were fabricated using solutions of CS (1% w/v) in 2% acetic acid and 98% deionized water (v/v), PLLA (10% (w/v) and PVAc (10% w/v)) (both in chloroform) and PEI–biotin (20% w/v) in water, which were mixed at the volume ratio (using the above solutions) of CS/PLLA/PVAc/PEI–biotin (10/2.5/2.5/0.1). The mixture was thoroughly mixed using a vortex mixer for 15 min (volume approximately 12 ml). The blended solution was then transferred into silicon moulds with dimensions of diameter = 17 mm and height = 2 mm, frozen in liquid nitrogen and lyophilised while maintaining a temperature of −20 °C until no solvent remained, yielding scaffold cylinders.Combination of polymer scaffold and PLGA-NPs
PLGA-NPs was prepared at 10 mg ml−1 in water; the polymer scaffold (17 mm × 2 mm) was incubated with the PLGA-NP suspension (200 μl) for 10 min with gently shaking. Scaffolds were removed from the PLGA-NP solution and placed in water for 2 × 10 minutes and lyophilised.Soluble collagen quantification
MC3T3-E1 cells were plated at a density of 20000 cells per cm2 in DMEM, 10% FBS and 1% penicillin/streptomycin. The following day, medium was removed and cell culture inserts were placed above the cells. Medium containing DMEM, 10% FBS and 1% penicillin/streptomycin, 2 mM glycerol 2-phosphate was added to the well compartment while medium supplemented with free ascorbate at a concentration of 1, 5, 25, 50 μg ml−1, containing NP or medium alone was added to the insert. The concentration of NP added to cell culture was 1 mg NP per ml medium. The medium containing free ascorbate, NP-AA2P or NP-blank was changed every 2–3 days. After 7 days of culture, cells were washed with DPBS and lysed for collagen and DNA quantification. After overnight lysis with 0.5 M acetic acid containing 0.1 mg pepsin, total collagen content was assessed using the Sircol™ soluble collagen assay according to the manufacturers protocol. Briefly, 100 μl sample was incubated with 1 ml Sircol Dye Reagent on a mechanical shaker for 30 minutes. Subsequently, samples were centrifuged at 12000 rpm for 10 minutes and the supernatant was removed. Samples were washed with ice-cold “Acid-Wash Reagent” and centrifugation was repeated. After removing any liquid residue, samples were dissolved in the “Alkali Reagent” and the absorbance was read at 560 nm. Concentration of collagen in samples was determined from the equation generated from a standard curve. Results were normalised to DNA content.
DNA quantification
DNA in samples lysed in CelLytic™ M were quantified using Quant-iT PicoGreen dsDNA Assay Kit according to the manufactures protocol. Briefly, samples were incubated with the PicoGreen reagent for 5 minutes and fluorescence was measured (480 nm/520 nm). DNA levels were determined using the standard curve.Tubule assay
Human umbilical cords were obtained, following signed consent, from healthy mothers after normal, full-term deliveries from the Princess Anne Hospital, Southampton, under ethical approval from the Southampton & South West Hampshire Local Research Ethics Committee (LREC 05/Q1702/102). HUVEC were isolated and cultured as described by Jaffe9 with minor modifications. In brief, human umbilical cord blood vein cells (HUVEC) were isolated using collagenase A and expanded in M199 medium containing ECGS/H, 10% FBS and 1% penicillin/streptomycin. For the tubule assay 40000 cells per cm2 were seeded in tissue culture plates covered with 3% LAPONITE®-fibronectin hydrogel (n = 3 per group). Cells were incubated for 18 hours in medium containing 40 ng ml−1 FGF-2 and with the addition of VEGF added to medium or released into the medium from VEGF-NPs at 37 °C, 5% CO2 in a humidified atmosphere. No addition of VEGF or supernatant from NP-blank was used as negative controls. Medium from VEGF-NPs and blank-NPs was collected after 6 hours incubation of the NP's at 37 °C followed by centrifugation at 12000 rpm. After incubation, cell cultures were imaged using an AxioVert200 Zeiss microscope and images were processed using Angiogenesis Analyzer task in ImageJ v2.0.0 to quantify total branching length. Four different areas from each sample were imaged and analysed.
Chorioallantoic membrane (CAM) assay
Animal procedures were carried out in accordance with the guidelines and regulations included in the Animals (Scientific Procedures) Act 1986, UK and chick embryo chorioallantoic membrane experimental protocols were approved and performed under Home Office Approval UK (Project licence – PPL P3E01C456) approved at the University of Southampton. The eggs were incubated horizontally for 10 days at 37 °C in a 60% humidified atmosphere, using a Hatchmaster incubator (Brinsea, UK) with one hour scheduled rotation. At day 10-post fertilisation, a scalpel blade was used to make an approximate 0.5cm2 square incision on the eggshell under sterile conditions and the eggshell fragment was removed to access the CAM beneath. Scaffolds containing blank-NPs or VEGF-NPs were sterilised under UV for 40 minutes and incubated in 30% antibiotic–antimitotic solution for 30 minutes. After extensive washing with DPBS, scaffolds were placed on the CAM of the chick embryo. For controls 30 μl DPBS with 25 ng and 100 ng VEGF or without VEGF was placed on the CAM of the chick embryo. Six to eight eggs were used for each experimental condition. The incision in eggshell was sealed with sterile parafilm secured with autoclave tape. After incubation without rotation for 8 days, CAM containing scaffolds and from control groups, were isolated and the gestational process was terminated following Home Office specific guidelines.
Chalkley point-overlap morphometry
Isolated CAMs from chick embryos were fixed in 4% PFA for one hour and vascularisation evaluated using Chalkley point-overlap morphometry (Chalkley score) with a Stemi 2000-C Zeiss microscope at 10 cm distance of sample from objective and 1.25× magnification. Five areas having the most concentrated vascular structures on each CAM were assessed and the count of the highest collision points of Chalkley graticule and vasculature on CAM was recorded individually as score.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council (Advanced Grant ADREEM ERC-2013-340469) and Biotechnology and Biological Sciences Research Council (BB/L00609X/1).Notes and references
- E. S. Place, N. D. Evans and M. M. Stevens, Nat. Mater., 2009, 8, 457–470 CrossRef PubMed
.
- P. H. Liebesny, S. Byun, H.-H. Hung, J. R. Pancoast, K. A. Mroszczyk, W. T. Young, R. T. Lee, D. D. Frisbie, J. D. Kisiday and A. J. Grodzinsky, Tissue Eng., Part A, 2016, 22, 917–927 CrossRef PubMed
- A. J. Lozano, H. J. Cestero and K. E. Salyer, J. Biomed. Mater. Res., Part A, 1976, 10, 545–548 CrossRef PubMed
- K. M. C. Chow and A. B. M. Rabie, Cleft Palate Craniofac. J., 2009, 37, 385–394 CrossRef
- B. Li, H. Wang, G. Zhou, J. Zhang, X. Su, Z. Huang, Q. Li, Z. Wu and G. Qiu, RSC Adv., 2017, 7, 4253–4259 RSC
- A. B. M. Rabie, Connect. Tissue Res., 2009, 36, 337–345 CrossRef
- D. Gothard, E. L. Smith, J. M. Kanczler, H. Rashidi, O. Qutachi, J. Henstock, M. Rotherham, A. El Haj, K. M. Shakesheff and R. O. C. Oreffo, Eur. Cells Mater., 2014, 28, 166–208 CrossRef PubMed
- Y. Liu, A. D. Berendsen, S. Jia, S. Lotinun, R. Baron, N. Ferrara and B. R. Olsen, J. Clin. Invest., 2012, 122, 3101–3113 CrossRef PubMed
- E. A. Jaffe, R. L. Nachman, C. G. Becker and C. R. Minick, J. Clin. Invest., 1973, 52, 2745–2756 CrossRef PubMed
- J. M. Kanczler and R. O. Oreffo, Eur. Cells Mater., 2008, 100–114 CrossRef
- R. T. Franceschi and B. S. Iyer, J. Bone Miner. Res., 1992, 7, 235–246 CrossRef PubMed
- B. L. Lyons and R. I. Schwarz, Nucleic Acids Res., 1984, 12, 2569–2579 CrossRef PubMed
- R. A. Jain, Biomaterials, 2000, 21, 2475–2490 CrossRef PubMed
- T. Govender, S. Stolnik, M. Garnett, L. Illum and S. Davis, J. Controlled Release, 1999, 57, 171–185 CrossRef PubMed
- H. Murakami, M. Kobayashi and H. Takeuchi, Int. J. Pharm., 1999, 143–152 CrossRef
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 329–347 CrossRef
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef PubMed
- M. Cegnar, A. Premzl, V. Zavašnik-Bergant, J. Kristl and J. Kos, Exp. Cell Res., 2004, 301, 223–231 CrossRef PubMed
- N. Obermajer, M. Cegnar, J. Kos and J. Kristl, J. Controlled Release, 2007, 18–26 Search PubMed
- J. Davda and V. Labhasetwar, Int. J. Pharm., 2002, 233, 51–59 CrossRef PubMed
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef PubMed
- J. S. Golub, Y.-T. Kim, C. L. Duvall, R. V. Bellamkonda, D. Gupta, A. S. Lin, D. Weiss, W. R. Taylor and R. E. Guldberg, Am. J. Physiol. Heart Circ. Physiol., 2010, 298, H1959–H1965 CrossRef PubMed
- L. E. Strong, S. N. Dahotre and J. L. West, J. Controlled Release, 2014, 178, 63–68 CrossRef PubMed
- G. Huang, J. Gao, Z. Hu, J. V. St John, B. C. Ponder and D. Moro, J. Controlled Release, 2004, 94, 303–311 CrossRef PubMed
- K. Nazemi, F. Moztarzadeh, N. Jalali, S. Asgari and M. Mozafari, BioMed Res. Int., 2014, 2014, 1–9 CrossRef PubMed
- K. Nazemi, P. Azadpour, F. Moztarzadeh, A. M. Urbanska and M. Mozafari, Mater. Lett., 2015, 138, 16–20 CrossRef
- X. Jiang, H. Lin, D. Jiang, G. Xu, X. Fang, L. He, M. Xu, B. Tang, Z. Wang, D. Cui, F. Chen and H. Geng, Sci. Rep., 2016, 6, 20784 CrossRef PubMed
- L. Li, G. Zhou, Y. Wang, G. Yang, S. Ding and S. Zhou, Biomaterials, 2015, 37, 218–229 CrossRef PubMed
- F. Khan, J. O. Smith, J. M. Kanczler, R. S. Tare, R. O. C. Oreffo and M. Bradley, Adv. Funct. Mater., 2013, 23, 2850–2862 CrossRef
- S. Takamizawa, Cell Biol. Int., 2004, 28, 255–265 CrossRef PubMed
- O. Fain, Jt., Bone, Spine, 2005, 72, 124–128 CrossRef PubMed
- W. Xing, S. Pourteymoor and S. Mohan, Physiol. Genomics, 2011, 43, 749–757 CrossRef PubMed
- F. Langenbach and J. Handschel, Stem Cell Res. Ther., 2013, 4, 117 CrossRef PubMed
- N. Jaiswal, S. E. Haynesworth, A. I. Caplan and S. P. Bruder, J. Cell. Biochem., 1997, 64, 295–312 CrossRef PubMed
- C. R. Ozawa, A. Banfi, N. L. Glazer, G. Thurston, M. L. Springer, P. E. Kraft, D. M. McDonald and H. M. Blau, J. Clin. Invest., 2004, 113, 516–527 CrossRef PubMed
- A. Faulkner, R. Purcell, A. Hibbert, S. Latham, S. Thomson, W. L. Hall, C. Wheeler-Jones and D. Bishop-Bailey, BMC Cell Biol., 2014, 15, 41 CrossRef PubMed
- P. Losi, E. Briganti, C. Errico, A. Lisella, E. Sanguinetti, F. Chiellini and G. Soldani, Acta Biomater., 2013, 9, 7814–7821 CrossRef PubMed
- Y.-I. Chung, S.-K. Kim, Y.-K. Lee, S.-J. Park, K.-O. Cho, S. H. Yuk, G. Tae and Y. H. Kim, J. Controlled Release, 2010, 143, 282–289 CrossRef PubMed
- A. Hernández, R. Reyes, E. Sánchez, M. Rodríguez Évora, A. Delgado and C. Évora, J. Biomed. Mater. Res., Part A, 2012, 100, 2382–2391 Search PubMed
- M. R. Hausman, M. B. Schaffler and R. J. Majeska, Bone, 2001, 29, 560–564 CrossRef PubMed
- M. P. Akhter, J. M. Lappe, K. M. Davies and R. R. Recker, Bone, 2007, 41, 111–116 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb00474a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |