
Covalent functionalization of graphene oxide with d-mannose: evaluating the hemolytic effect and protein corona formation†
 *a
Carlos H. Z.
Martins,
a
Lidiane S.
Franqui,
bc
Diego Stéfani T.
Martinez
*abc
and
Oswaldo L.
Alves
*a
*a
Carlos H. Z.
Martins,
a
Lidiane S.
Franqui,
bc
Diego Stéfani T.
Martinez
*abc
and
Oswaldo L.
Alves
*a
Abstract
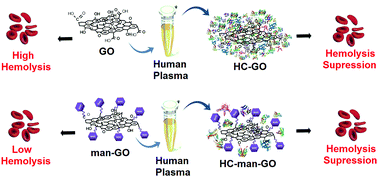
In this work, graphene oxide (GO) was covalently functionalized with D-mannose (man-GO) using mannosylated ethylenediamine. XPS (C1s and N1s) confirmed the functionalization of GO through the binding energies at 288.2 eV and 399.8 eV, respectively, which are attributed to the amide bond. ATR-FTIR spectroscopy showed an increase in the amine bond intensity, at 1625 cm−1 (stretching C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), after the functionalization step. Furthermore, the man-GO toxicity to human red blood cells (hemolysis) and its nanobiointeractions with human plasma proteins (hard corona formation) were evaluated. The mannosylation of GO drastically reduced its toxicity to red blood cells. SDS-PAGE analysis showed that the mannosylation process of GO also drastically reduced the amount of the proteins in the hard corona. Additionally, proteomics analysis by LC–MS/MS revealed 109 proteins in the composition of the man-GO hard corona. Finally, this work contributes to future biomedical applications of graphene-based materials functionalized with active biomolecules.
O), after the functionalization step. Furthermore, the man-GO toxicity to human red blood cells (hemolysis) and its nanobiointeractions with human plasma proteins (hard corona formation) were evaluated. The mannosylation of GO drastically reduced its toxicity to red blood cells. SDS-PAGE analysis showed that the mannosylation process of GO also drastically reduced the amount of the proteins in the hard corona. Additionally, proteomics analysis by LC–MS/MS revealed 109 proteins in the composition of the man-GO hard corona. Finally, this work contributes to future biomedical applications of graphene-based materials functionalized with active biomolecules.
- This article is part of the themed collections: Materials and Nano Research in Brazil and International Year of the Periodic Table : Low Dimensional Carbon Systems
 Please wait while we load your content...
Please wait while we load your content...

