
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stable DHLA–PEG capped PbS quantum dots: from synthesis to near-infrared biomedical imaging†
F.
Zamberlan
 *ab,
L.
Turyanska
*bc,
A.
Patanè
c,
Z.
Liu
a,
H. E. L.
Williams
a,
M. W.
Fay
d,
P. A.
Clarke
e,
Y.
Imamura
f,
T.
Jin
f,
T. D.
Bradshaw
g,
N. R.
Thomas
a and
A. M.
Grabowska
e
*ab,
L.
Turyanska
*bc,
A.
Patanè
c,
Z.
Liu
a,
H. E. L.
Williams
a,
M. W.
Fay
d,
P. A.
Clarke
e,
Y.
Imamura
f,
T.
Jin
f,
T. D.
Bradshaw
g,
N. R.
Thomas
a and
A. M.
Grabowska
e
aCentre for Biomolecular Sciences, School of Chemistry, University of Nottingham, University Park, NG72RD, UK. E-mail: Francesco.Zamberlan@nottingham.ac.uk
bSchool of Chemistry, University of Lincoln, LN6 7DL, UK
cSchool of Physics and Astronomy, University of Nottingham, University Park, NG72RD, UK. E-mail: Lyudmila.Turyanska@nottingham.ac.uk
dNanoscale and Microscale Research Centre, University of Nottingham, University Park, NG72RD, UK
eCancer Biology, Division of Cancer and Stem Cells, School of Medicine, University of Nottingham, Queen's Medical Centre, NG7 2UH, UK
fLaboratory for Nano-Bio Probes, Quantitative Biology Centre, RIKEN, Osaka, 565-0874, Japan
gCentre for Biomolecular Sciences, School of Pharmacy, University of Nottingham, University Park, NG72RD, UK
First published on 12th January 2018
Abstract
The short shelf-life of water-soluble quantum dots (QDs) due to colloidal instability represents a major drawback to their exploitation. This work examines the colloidal stability of PbS nanoparticles capped with dihydrolipoic acid–polyethylene glycol (DHLA–PEG) ligands terminated with functional groups such as –NH2, –COOH, OMe and –N3. and their application for in vivo imaging. We prove a mechanism of colloidal instability and develop a strategy to produce for the first time stable PEG-capped PbS quantum dots with high quantum yield and optical emission in the first and the second near-infrared (NIR) windows of low absorption of biological tissues. The NIR imaging of in vivo biodistribution is demonstrated at wavelengths >1000 nm, with benefits of reduced tissue absorption and light scattering. The stability, biocompatibility and potential for further QD functionalization open up realistic prospects for non-invasive bioimaging applications.
The stability of colloidal nanoparticle solutions in physiological solvents is a key prerequisite for their applications in bioimaging and nanomedicine.1–3 Various nanomaterial systems have been explored for medical imaging, including metal nanoparticles,4,5 upconverting nanoparticles6 and semiconductor quantum dots.7 Of particular interest is the development of biocompatible colloidal quantum dots (QDs) with optical emission tuneable in the near-infrared (NIR) wavelength range of reduced light scattering and low absorption of biological tissues,8–10 which can be achieved with IV–VI-based semiconductor materials (e.g. PbS, PbSe).9,11 The important role of surface ligands for controlling and designing functional nanomaterials has been widely acknowledged.12,13 It has also been shown that the efficiency of ligand binding to the surface of a nanocrystal has a strong influence on QD polydispersity, quantum yield and photostability.14,15 Polymer-based capping ligands that are biologically inert, e.g. polyethylene glycol (PEG),3,16,17 have been successfully used to produce a range of colloidal nanomaterials, from metal nanoparticles to semiconductor QDs.13,16–19 However, the development of capping ligands that can effectively passivate the surface of a nanocrystal, provide longevity and colloidal stability, do not alter its optical properties and allow controllable attachment of functional molecules remains a challenge.13,20 On the other hand, the modular attachment of PEG to dihydrolipoic acid (DHLA) as the anchoring moiety of the ligand has been shown to improve the passivation of nanoparticles, such as AuNP and CdSe–ZnS QDs.21–24 Despite promising optical and electronic properties, the technological exploitation of these materials has stalled due to their short shelf-life caused by colloidal instability.
Here we report the development of nanoparticles that combine the beneficial biocompatibility and stability offered by DHLA–PEG-ligands with the tuneability of the emission of PbS nanocrystals in the NIR wavelength range of low absorption of biological tissues. We report on the stability of DHLA–PEG ligands and their utilization for surface passivation of quantum dots. We investigate the reasons for previously reported colloidal instability and short shelf-life of colloidal solutions of thiol-linked, PEG-capped nanoparticles. We report time-dependent nuclear magnetic resonance (NMR) studies of ligand molecules and QDs, and suggest a strategy for the synthesis of stable nanoparticle solutions. The DHLA–PEG ligands, modified with functional groups including −OMe, −NH2, −COOH and −N3, are used to synthesize colloidal PbS QDs in aqueous solution with high quantum yield (∼30%) and with room temperature photoluminescence (PL) tuneable by the QD core size in the NIR wavelength range, hence enabling enhanced light penetration through deep tissues and in vivo NIR imaging of QD biodistribution in mice. These results open up prospects for use of PbS–DHLA–PEG QDs in biomedical applications where functional groups can be used for the controllable attachment of targeting molecules, such as peptides, small proteins or antibodies.
The ligands used in this work are composed of three parts: DHLA, a PEG400 linker, and a functional group, R*, such as −OMe, −NH2, −COOH and −N3,22,25 corresponding to ligand molecules LOMe, LNH2, LCOOH and LN3, respectively. PEG400 is a hydrophilic molecule that provides water solubility, pH stability and biocompatibility of the QDs, and can be easily changed for a different chain length from PEG200 to PEG2000. The stability and aging of ligand molecules has been assessed by NMR time-course (Fig. 1 and ESI,† SI2). Our 1H-NMR studies indicate that for the LNH2 and LN3 molecules, the spontaneous oxidation of the DHLA and reformation of the disulfide bond of its parent compound lipoic acid (LA) takes place with ∼70% conversion over 48 h (Fig. 1a). A clear resurgence of the proton signals at ∼2.45 ppm (Hb*) and at ∼3.18 ppm (Ha*/a′*) (see Fig. 1b) is observed in LNH2, which are typical of the 5-membered dithiolane ring containing the disulfide bond. At the same time, the intensities of peaks at 2.71 ppm (Ha/a′) and 2.94 ppm (Hc), characteristic of DHLA, decrease. The change of intensities is described by the first order kinetic regime with comparable time constants (inset in Fig. 1b), as expected for the interconversion reaction where DHLA is oxidized to LA. These results indicate the inherent instability of LNH2 and LN3, which is likely due to the interaction between the amine moiety and the thiols.26,27 On the other hand, LOMe and LCOOH are stable and do not undergo spontaneous oxidation (Fig. 1c and ESI,† SI2) over the same observation period. The formation of the disulfide bond prevents the ligand binding to the nanoparticle surface, thus affecting the colloidal stability of QDs.
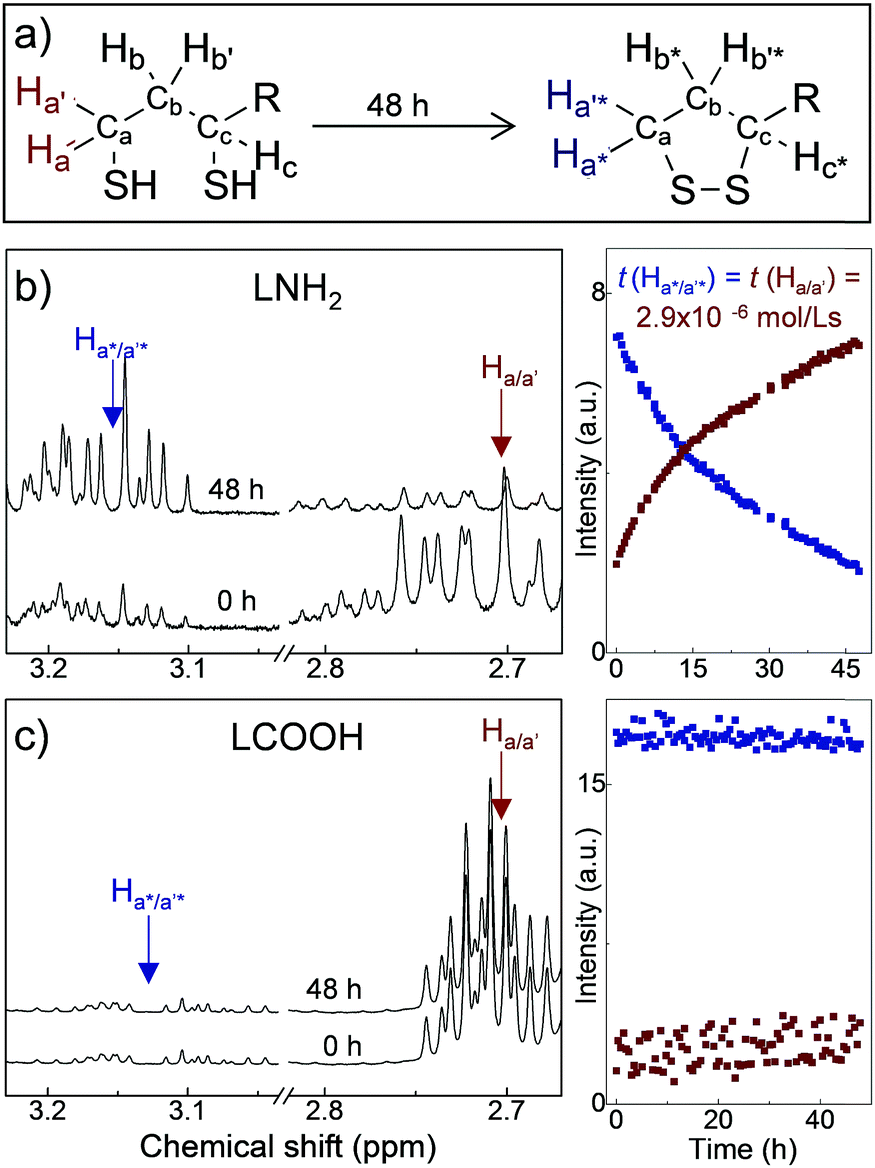 | ||
| Fig. 1 (a) Reaction scheme of the oxidation of the dithiols to the dithiolane ring; R is the remaining part of lipoic acid–PEG400-terminating group. Representative 1H-NMR spectra of LNH2 (b) and LCOOH (c) for freshly synthesised ligands and following 48 h storage, with corresponding time dependence of the intensities of selected resonances. | ||
Particular attention to the purification of the ligand mixture is paramount for the stability of aqueous QD solutions. During the ligand synthesis, the Staudinger reduction uses triphenyl phosphine (Ph3P), generating triphenylphosphine oxide (Ph3P(O)) as a by-product. The 31P NMR shows a clear peak at −29 ppm, confirming the presence of Ph3P(O) (ESI,† SI2). The Ph3P(O)/Ph3P present in the reaction mixture can compete with the DHLA–PEG as a QD capping molecule, thus altering the aqueous solubility of the nanocrystals.28
We have used these DHLA–PEG capping ligands to produce PbS quantum dots either in aqueous solution or in phosphate buffered saline (PBS) (Fig. 2a) with sizes tuneable by the molar ratio of Pb![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S.29 For the Pb:S ratio of 1:0.3, the QDs have average diameter dPbS = 3.2 ± 0.8 nm (Fig. 2b). The dark field TEM studies reveal the presence of a thick carbon-based amorphous layer with non-uniform depth surrounding the PbS nanocrystal. The inter-particle distance value of ≥2 nm indicates interlocking of the DHLA–PEG between neighboring nanocrystals on the surface.30,31 The average hydrodynamic diameter of the QDs measured using dynamic light scattering (DLS) is dQD = 11 ± 1 nm, which is close to our estimate dQD = dPbS + 2l ≈ 13 nm, where l ≈ 5 nm is the length of the extended ligand molecule. The QD solutions prepared with molar ratio of Pb:S = 1:0.3, have room temperature photoluminescence centered at λPL = 920 nm, which is independent of the type of ligand and/or ligand mixture used for surface passivation (Fig. 2c).
:S.29 For the Pb:S ratio of 1:0.3, the QDs have average diameter dPbS = 3.2 ± 0.8 nm (Fig. 2b). The dark field TEM studies reveal the presence of a thick carbon-based amorphous layer with non-uniform depth surrounding the PbS nanocrystal. The inter-particle distance value of ≥2 nm indicates interlocking of the DHLA–PEG between neighboring nanocrystals on the surface.30,31 The average hydrodynamic diameter of the QDs measured using dynamic light scattering (DLS) is dQD = 11 ± 1 nm, which is close to our estimate dQD = dPbS + 2l ≈ 13 nm, where l ≈ 5 nm is the length of the extended ligand molecule. The QD solutions prepared with molar ratio of Pb:S = 1:0.3, have room temperature photoluminescence centered at λPL = 920 nm, which is independent of the type of ligand and/or ligand mixture used for surface passivation (Fig. 2c).
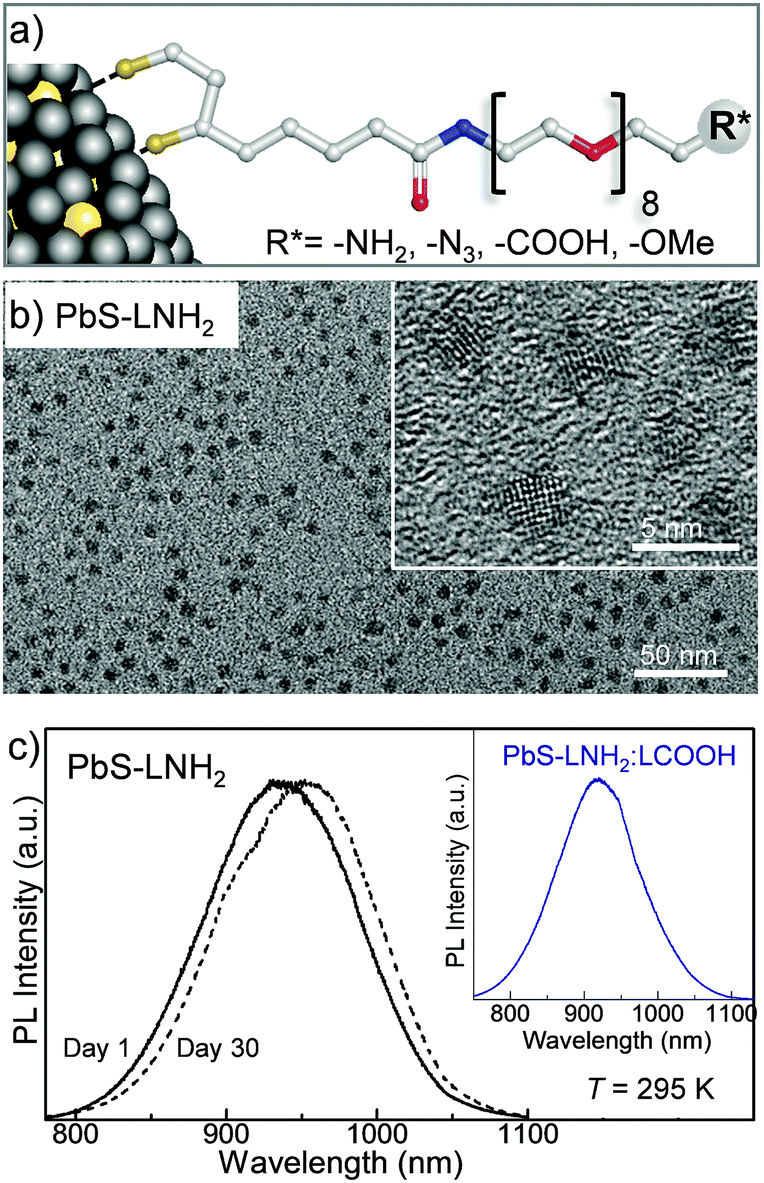 | ||
| Fig. 2 (a) Schematic representation of a nanoparticle capped with DHLA–PEG-based ligands. (b) TEM and (inset) HR TEM images of PbS QDs synthesized with LNH2 capping ligands and molar ratio Pb:S = 1:0.3. (c) Room temperature photoluminescence (PL) spectra of freshly synthesized (solid line) and 30-days-old (dashed line) PbS QDs (Pb:S = 1:0.3). Inset: Room temperature PL spectrum of PbS QDs with molar ration of Pb:S = 1:0.3, synthesized with a mixture of LNH2 and LCOOH ligands in 1:3 molar ratio. | ||
By varying the Pb:S ratio from 1:0.3 to 1:0.6 we produce quantum dots with a diameter ranging from dPbS ≈ 3.2 nm to 5.5 nm, with corresponding PL emission tuneable in the NIR range from 920 nm to 1220 nm, respectively (Fig. 3a). The PL peak position and intensity are comparable for all ligands used, irrespective of the specific terminating group (−OMe, −NH2, −COOH and −N3) and only small time-dependent shift is observed due to Ostwald ripening (Fig. 2c). All PbS QD solutions have quantum yields of about 30% as measured using the integrated sphere method (Edinburg Instrument spectrometer FLS980 equipped with 120 mm diameter integrated sphere), which is higher than that previously reported for other types of PbS QDs.32 We note that stable colloidal solutions of PbS QDs can also be produced with mixed ligands, e.g. LNH2:LCOOH in a 3:1 molar ratio (inset of Fig. 2c). The mixing of the ligands has a potential benefit for the controlled attachment of different molecules. This in turn could allow greater selectivity, e.g. attachment of complementary targeting molecules. Also, the control of the ratio of functional groups on the QD surface allows tuneability of the surface charge from positive for PbS–LNH2 (ζ-potential ∼ 2.6 ± 3.2 mV) to negative for PbS–LCOOH QD (ζ-potential ∼ −41.3 ± 5.5 mV).
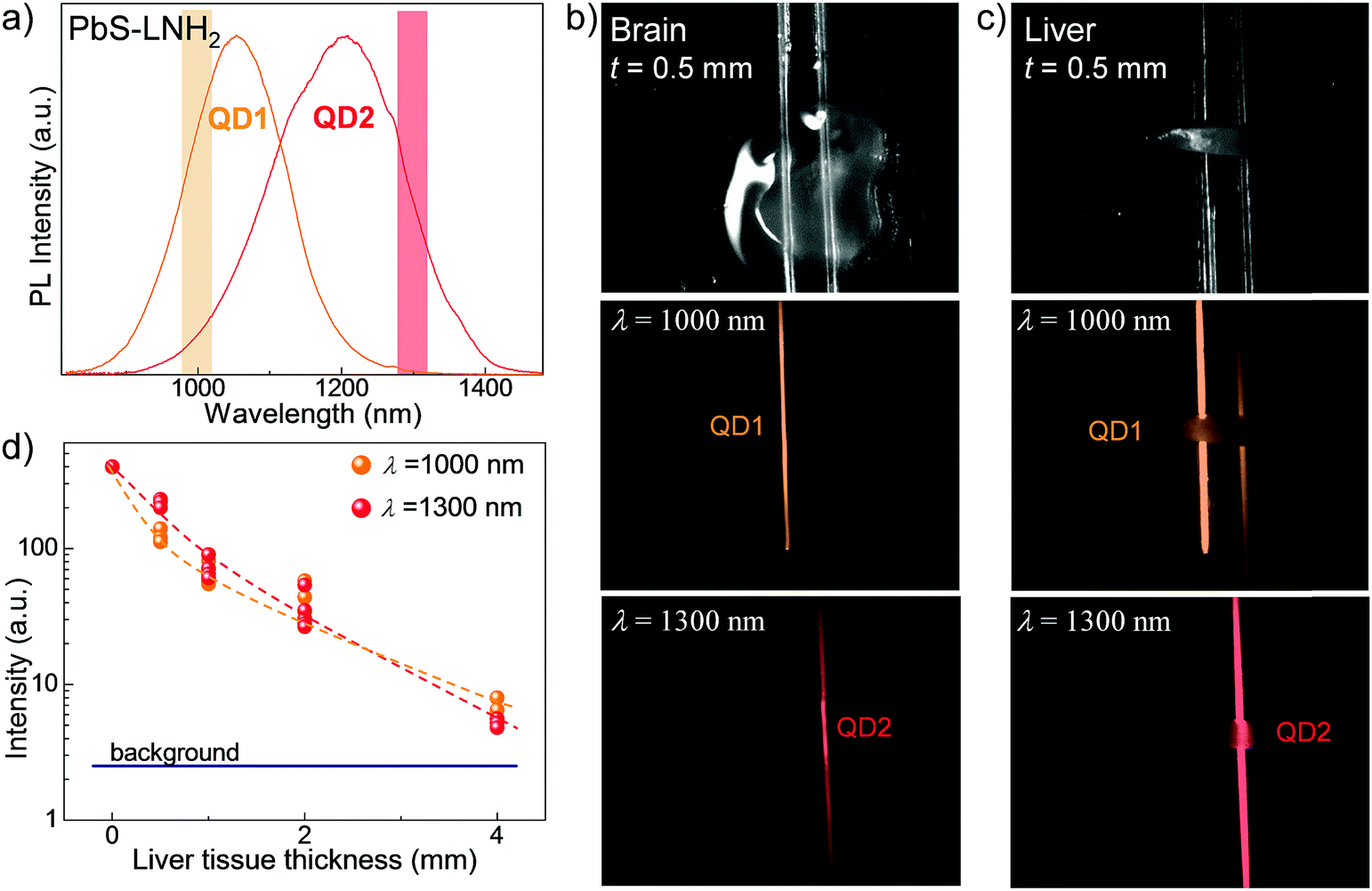 | ||
| Fig. 3 (a) Room temperature PL spectra of PbS–LNH2 nanocrystals QD1 (orange spectrum) and QD2 (red spectrum). The highlighted areas indicate the wavelength range where PL images through the tissues are acquired. Optical images and corresponding PL images of QD loaded capillary tubes overlaid with (b) brain and (c) liver tissue slices of thickness t = 0.5 mm. PL images were acquired with excitation wavelength λex = 670 nm and bandpass filters at (1000 ± 25) nm and at (1300 ± 25) nm. (d) PL intensity measured through the liver tissues versus the tissue thickness at λ = 1000 nm and λ = 1300 nm. Dashed lines are guide to the eye. | ||
The QD solutions prepared using freshly synthesized ligands have long-term colloidal stability (stored at T = 4 °C under nitrogen atmosphere) over a period of at least 9 months. 1H-NMR studies of these QD solutions show a clear resonances at 2.71 ppm (Ha/a′) and 2.94 ppm (Hc), characteristic for DHLA. In stark contrast, when ligands were stored prior to the nanocrystal synthesis, QD solutions precipitated within <10 days and the 1H-NMR spectra of the supernatant confirmed the formation of the dithiolane ring on the ligand molecules. Hence the instability is due to lack of surface passivation by the unreactive oxidized ligand molecules.
The potential of these QDs for bioimaging applications was assessed by studies of the QD NIR emission penetration through biological tissues. For these studies we selected the QD solutions with PL at λ > 1000 nm to benefit from low absorption of biological tissues and lower light scattering, thus achieving deeper tissue penetration of the emitted light. To account for the composition of different tissues, we prepared mouse brain and liver tissue slices with thicknesses ranging from t = 0.2 mm to 4 mm. The slices were placed over two glass capillaries of diameter d = 1 mm containing PbS–LNH2 solutions with PL emission of comparable intensity centered at λPL ∼ 1050 nm (QD1) and at λPL ∼ 1220 nm (QD2) (Fig. 3a). Fig. 3b and c illustrate representative optical images of 0.5 mm thick brain and liver slices, respectively, placed over the capillaries. The corresponding PL maps were acquired at λ = 1000 ± 25 nm (middle images) and at 1300 ± 25 nm (bottom images). They demonstrate that the NIR fluorescence of the QDs can be detected through all studied tissues.
However, the observed decrease of light intensity through tissues is significantly greater for high-blood-content tissues, such as liver, compared to brain. For example, for brain tissues of thickness t = 0.5 mm, no measurable decrease of optical signal was detected compared to capillary only. For the same thickness of liver tissue, about 5-times decreased intensity was observed at λ = 1000 ± 25 nm and 2-times decreased intensity at λ = 1300 ± 25 nm (Fig. 3d). The use of longer wavelengths enables better imaging through liver, with ∼1.5 times higher resolution achieved for all tissue thicknesses (Fig. 3c and ESI,† SI3) due to reduced scattering.8,33 We also note that a signal to noise ratio of ≥3 was achieved for liver tissue of thicknesses up to 4 mm. The observed signal penetration depth confirms the suitability of these QDs for non-invasive imaging.
In order to guide the in vivo studies, we assessed the cytotoxicity of these QDs in vitro by MTT assay. Prior to cell culture and in vivo studies, we dialyse the QD solutions against PBS and lower the pH. We note that this process does not alter the colloidal stability and photostability of the QDs. Previously, PbS QDs were reported to induce greater toxicity in tumorigenic cell lines compared to normal cells.29,34 We selected three human derived carcinoma cell lines: MDA-MB-231 and MCF-7 breast, and HCT-116 colorectal cells. Assessment after 72 h exposure of cells to PbS QDs demonstrated that in all three cell lines, neither cell viability nor cell growth were compromised at PbS QD concentrations ≤20 μg mL−1 (see ESI,† SI3). The acute toxicity studies over a period of 30 days have not revealed any QD-induced toxic effects in mice.34 No indication of damage to the organs/tissues was found in histological examination of internal organs at the end of the study, confirming the stability of QDs in vivo and the absence of release of heavy metal ions from the QDs.
The in vivo biodistribution of QDs was observed using NIR fluorescence. Mice were injected via tail vein with 30 μL of 0.2 mM PbS QD solution, corresponding to a final concentration of approximately 10 μg mL−1 (2-times lower than the GI50 value). Fig. 4a illustrates typical NIR images (λ > 1000 nm) of PbS QDs (LNH2) in blood vessels in a mouse head immediately after the injection. The fluorescence in the brain vessels fades within a few minutes and the emission in not detectable upon inspection of the brain ex vivo 48 h after injection, indicating that these QDs do not pass the blood–brain barrier.
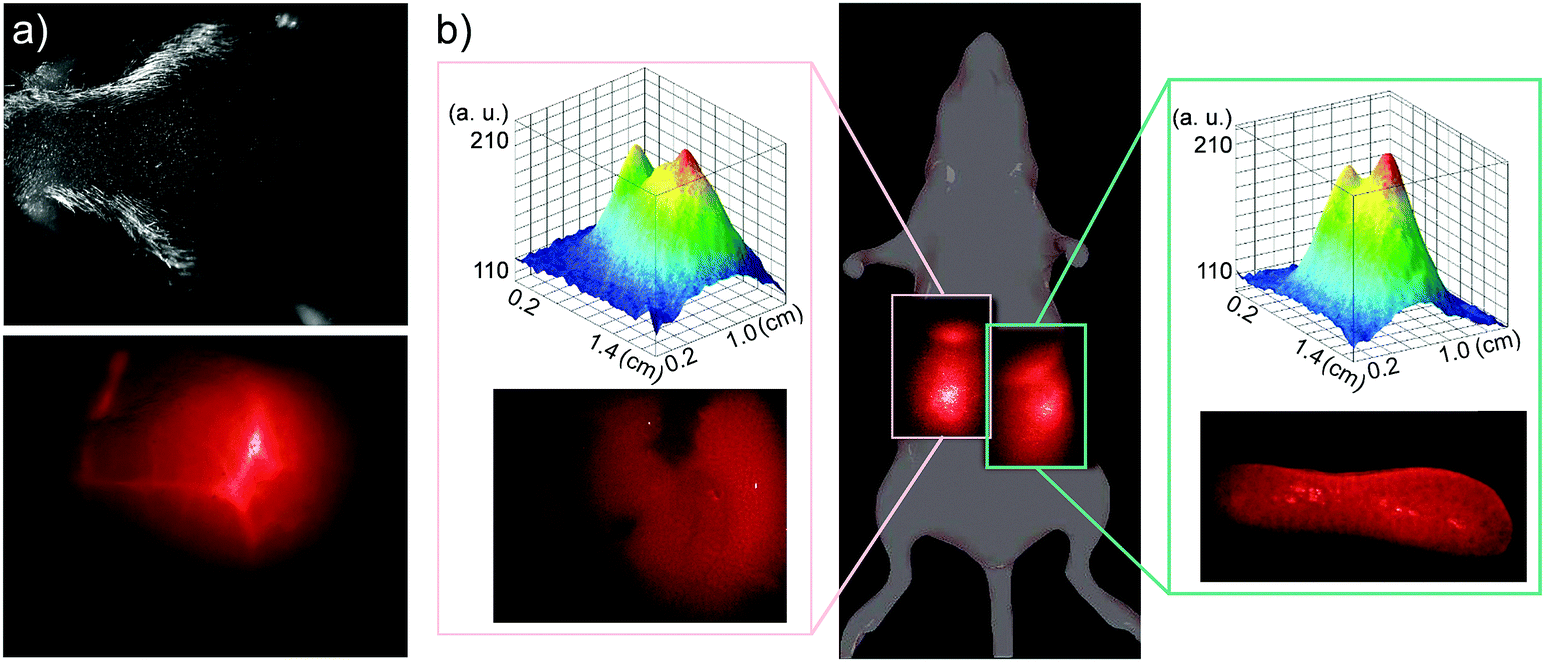 | ||
| Fig. 4 (a) Photograph of a mouse head and a corresponding NIR fluorescence image of blood vessels in the head immediately after tail vein injection of LNH2 capped QDs (PL centred at 1050 nm). (b) Whole body and corresponding intensity surface, and ex vivo NIR fluorescence images of liver (left inset) and spleen (right inset) of a mouse 24 h after tail vein injection of LNH2 capped QDs (PL centred at 1220 nm). NIR images were acquired with excitation laser at λex = 670 nm (P = 10 W m−2), InGaAs CMOS camera and a long pass filter at 1000 nm. | ||
Within 1 hour, measurable NIR fluorescence was observed in the areas of liver and spleen. Fig. 4b illustrates a typical biodistribution of the PbS QDs, as detected with NIR fluorescence 24 hours after the QD injection. The intensities of the fluorescence signal observed in liver and in spleen are comparable when measured in vivo (see top insets in Fig. 4b) and ex vivo in the corresponding organs (see lower insets of Fig. 4b). In agreement with our tissue penetration studies, the in vivo fluorescence intensity is about 2-times lower compared to the ex vivo value. We note that, as expected, NIR fluorescence was only detected in QD injected animals (see ESI,† SI3). Following 48 h exposure, the intensity of the optical signal from both liver and spleen decreased by a factor of ∼3 compared to that measured after 24 h exposure. We attribute the observed decrease of fluorescence to gradual clearance of QDs from the body. Indeed in some mice, significant fluorescence was observed in the bladder area, indicating that QDs are cleared from the body via the urinary tract. There is also some indication that after 48 h some QDs might be present in lymph nodes, as was also reported previously.33,35 We exclude the possibility that the decrease in fluorescence intensity is due to interactions of QDs with proteins in blood/serum (e.g. with albumin), as we prepare our QDs with excess ligands to ensure efficient passivation of the surface in order to eliminate such interactions. Also, even in the case of QD binding to proteins in serum, the formed complexes are reported to be both soluble and photostable.36
We note that over the duration of the study, the QDs were well tolerated by mice. No distress or noticeable physiological changes were observed. Also, mice organs (liver, spleen, brain, kidneys, lungs and heart) were inspected after 24 h and after 48 h, and no visible morphological changes were observed. Ex vivo, measurable fluorescence was detected only in spleen and liver (lower insets in Fig. 4b). Importantly, no noticeable fluorescence was detected in kidneys, indicating that QDs do not accumulate and are well cleared via the kidneys. No noticeable changes in QD biodistribution and in fluorescence intensity were observed for QDs capped with ligands terminated with different functional groups.
In our preliminary studies, we have successfully attached folic acid and cyclic RGD peptides, as cellular targeting ligands, using –NH2 and –N3 terminated ligands, respectively, and produced nanoparticles with these functionalized ligands. Further work is underway to explore the targeting efficiency of these functionalized nanocrystals in tumour bearing mice and to fully assess the potential of these QDs for bioimaging applications.
Conclusions
In conclusion, we have identified and addressed the causes that trigger the instability of colloidal nanoparticles capped with DHLA–PEG-based ligands, i.e. LOMe, LNH2, LN3, LCOOH or their mixtures. Also, we have successfully used these molecules to produce stable and biocompatible PbS QDs with optical emission tuneable in the near-infrared wavelength range. The QDs are well tolerated by mice in vivo and their emission can be imaged in the second NIR optical window. Our findings are transferable to other types of semiconductor and metal nanoparticles and offer potential for further functionalization of the nanocrystals for a wide range of biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Centre for the Replacement, Refinement and Reduction of Animals in Research [grant number NC/L001861/1]; the Engineering and Physical Sciences Research Council [grant numbers EP/K503800/1, NC/L001861/1]; the Nanoscale and Microscale Research Centre; and The University of Nottingham.References
-
G. Palui, W. Wang, F. Aldeek and H. Mattoussi, Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Inc., 2013 Search PubMed
.
- K. Ulbrich, K. Holá, V. Šubr, A. Bakandritsos, J. Tuček and R. Zbořil, Chem. Rev., 2016, 116, 5338 CrossRef CAS PubMed
- J. Soo Suk, Q. Xua, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28 CrossRef PubMed
- J. Ando, T. Yano, K. Fujita and S. Kawata, Phys. Chem. Chem. Phys., 2013, 15, 13713 RSC
- L. Polavarapu, M. Manna and Q. Hua Xu, Nanoscale, 2011, 3, 429 RSC
- M. González-Béjar, L. Francés-Soriano and J. Pérez-Prieto, Front. Bioeng. Biotechnol., 2016, 4, 47 Search PubMed
- I. V. Martynenko, A. P. Litvin, F. Purcell-Milton, A. V. Baranov, A. V. Fedorov and Y. K. Gun’ko, J. Mater. Chem. B, 2017, 5, 6701 RSC
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710 CrossRef CAS PubMed
- J. Zhou, Y. Yang and C. Y. Zhang, Chem. Rev., 2015, 115, 11669 CrossRef CAS PubMed
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 10 CrossRef
- A. Sasaki, Y. Tsukasaki, A. Komatsuzaki, T. Sakata, H. Yasuda and T. Jin, Nanoscale, 2015, 7, 5115 RSC
- M. A. Walling, J. A. Novak and J. R. E. Shepard, Int. J. Mol. Sci., 2009, 10, 441 CrossRef CAS PubMed
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141 CrossRef CAS PubMed
- M. Green, J. Mater. Chem., 2010, 20, 5797 RSC
- Y. Zhang and A. Clapp, Sensors, 2011, 11, 11036 CrossRef PubMed
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 716 CrossRef PubMed
- G. Palui, F. Aldeek, W. Wang and H. Mattoussi, Chem. Soc. Rev., 2014, 44, 193 RSC
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346 CrossRef CAS PubMed
- W. Liu, M. Howarth, A. B. Greytak, Y. Zheng, D. G. Nocera, A. Y. Ting and M. G. Bawendi, J. Am. Chem. Soc., 2008, 130, 1274 CrossRef CAS PubMed
- A. M. Smith, H. Duan, A. M. Mohs and S. Nie, Adv. Drug Delivery Rev., 2008, 60, 1226 CrossRef CAS PubMed
- K. Susumu, H. T. Uyeda, I. L. Medintz, T. Pons, J. B. Delehanty and H. Mattoussi, J. Am. Chem. Soc., 2007, 129, 13987 CrossRef CAS PubMed
- K. Susumu, B. C. Mei and H. Mattoussi, Nat. Protoc., 2009, 4, 424 CrossRef CAS PubMed
- F. Aldeek, D. Hawkins, V. Palomo, M. Sa, G. Palui, P. E. Dawson, I. Alabugin and H. Mattoussi, J. Am. Chem. Soc., 2015, 137, 2704 CrossRef CAS PubMed
- N. Zhan, G. Palui, J. Merkl and H. Mattoussi, J. Am. Chem. Soc., 2016, 138, 3190 CrossRef CAS PubMed
- B. C. Mei, K. Susumu, I. L. Medintz and H. Mattoussi, Nat. Protoc., 2009, 4, 412 CrossRef CAS PubMed
- T. J. Wallace and A. Scriesheim, J. Org. Chem., 1961, 27, 1514 CrossRef
- T. J. Wallace, N. Jacobson and A. Schriesheim, Nature, 1964, 201, 609 CrossRef CAS
- C. B. Murray, D. J. Norriss and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS
- L. Turyanska, T. D. Bradshaw, J. Sharpe, M. Li, S. Mann, N. R. Thomas and A. Patane, Small, 2009, 5, 1738 CrossRef CAS PubMed
- Y. Justo, I. Moreels, K. Lambert and Z. Hens, Nanotechnology, 2010, 21, 295606 CrossRef PubMed
- J. Paczesny, M. Wolska-Pietkiewicz, I. Binkiewicz, M. Wadowska, Z. Wróbel, K. Matuła, W. Nogala, J. Lewiński and R. Hołyst, ACS Appl. Mater. Interfaces, 2016, 8, 13532 CAS
- M. Greben, A. Fucikova and J. Valenta, J. Appl. Phys., 2015, 117, 144306 CrossRef
- T. Jin and Y. Imamura, ECS J. Solid State Sci. Technol., 2016, 5, R3138 CrossRef CAS
- T. D. Bradshaw, J. Marc, A. Patane, P. Clarke, N. R. Thomas, M. Li, S. Mann and L. Turyanska, J. Mater. Chem. B, 2013, 1, 6254 RSC
- Y. Nakane, Y. Tsukasaki, T. Sakata, H. Yasuda and T. Jin, Chem. Commun., 2013, 49, 7584 RSC
- K. Hanaki, A. Momo, T. Oku, A. Komoto, S. Maenosono, Y. Yamaguchi and K. Yamamoto, Biochem. Biophys. Res. Commun., 2003, 302, 496 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb02912h |
| This journal is © The Royal Society of Chemistry 2018 |