
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interweaving metal–organic framework-templated Co–Ni layered double hydroxide nanocages with nanocellulose and carbon nanotubes to make flexible and foldable electrodes for energy storage devices†
Chao
Xu‡
 *ab,
Xueying
Kong‡
ab,
Shengyang
Zhou
b,
Bing
Zheng
*a,
Fengwei
Huo
a and
Maria
Strømme
*b
*ab,
Xueying
Kong‡
ab,
Shengyang
Zhou
b,
Bing
Zheng
*a,
Fengwei
Huo
a and
Maria
Strømme
*b
aKey Laboratory of Flexible Electronics (KLOFE), Institute of Advanced Materials (IAM), Nanjing Tech University (Nanjing Tech), 30 South Puzhu Road, Nanjing 211800, China. E-mail: chao.xu@angtrom.uu.se; iambzheng@njtech.edu.cn
bDivision of Nanotechnology and Functional Materials, Department of Engineering Sciences, Uppsala University, Uppsala SE-75121, Sweden. E-mail: maria.stromme@angstrom.uu.se
First published on 9th November 2018
Abstract
Metal–organic frameworks (MOFs) and nanocellulose represent emerging and traditional porous materials, respectively. The combination of these two materials in specific ways could generate novel nanomaterials with integrated advantages and versatile functionalities. This study outlines the development of hierarchical porous and conductive nanosheets based on zeolitic imidazolate framework-67 (ZIF-67, a Co-based MOF)-templated Co–Ni layered double hydroxide (LDH) nanocages, Cladophora cellulose (CC) nanofibers, and multi-walled carbon nanotubes (CNTs). The LDH–CC–CNT nanosheets can be used as flexible and foldable electrodes for energy storage devices (ESDs). The electrodes are associated with a high areal capacitance of up to 1979 mF cm−2 at a potential scan rate of 1 mV s−1. A flexible, foldable, and hybrid ESD is assembled from LDH–CC–CNT and CC–CNT electrodes with a PVA/KOH gel. The entire device has an areal capacitance of 168 mF cm−2 and an energy density of 0.6 mW h cm−3 (60 μW h cm−2), at a power density of 8.0 mW cm−3 (0.8 mW cm−2). These promising results demonstrate the potential of using MOFs and sustainable cellulose in flexible, foldable electronic energy storage devices.
Introduction
There is currently great interest in the development of high performance, thin, flexible, lightweight, environmentally friendly energy storage devices (ESDs).1–7 Supercapacitors (SCs) constitute an important class of electric energy storage systems that bridges the gap between conventional capacitors and batteries. Generally, SCs are classified as electrical double layer capacitors (EDLCs) and pseudocapacitors, depending on the charge storage mechanism and the active materials used.8 EDLCs store charges on the large internal surfaces of active materials such as activated carbon, carbon nanotubes (CNTs) and graphene.9 Pseudocapacitors rely on electrochemically active materials including certain transition metal oxides, hydroxides, and conducting polymers, for example, RuO2,10 MnO2,11 and polypyrrole,12 which store electrochemical energy faradaically via reversible redox reactions at the interface of the electrode and electrolyte and thus provide higher specific capacitances than EDLCs. In order to increase the energy density, two electrodes with different charge storage behaviours may be applied in the assembly of ESDs.13,14 For example, asymmetric supercapacitors pair one pseudocapacitive electrode and one capacitive electrode, e.g., MnO2 and activated carbon.15 Hybrid capacitors combine faradaic battery-type electrodes and capacitive electrodes, such as Ni(OH)2 and activated carbon.16 Because of their high power density, rapid charge/discharge rate, and long operational lifetime, a significant amount of effort has been devoted to the development of flexible SCs and hybrid devices to meet the growing demands of flexible electronics, for instance, foldable phones, roll-up displays, and wearable devices.17–19Metal–organic frameworks (MOFs) are a newly emerging family of functional materials with exceptionally high surface areas and well-defined pores that are constructed by coordination between metal ions or clusters and organic linkers.20,21 In addition to their well-studied applications in gas storage and separation,22 catalysis,23 drug delivery,24 and sensors,25 extensive research interest in MOFs has recently been triggered for the exploration of novel electrode materials for SCs.26–28 Their ordered structures and high surface areas result in fast diffusion of the electrolyte in microporous or mesoporous channels. In addition, transition metal ions or clusters in some MOFs are redox-active centers that can deliver electrochemical activity. Various pristine MOFs and MOF-derived materials have already been successfully applied as capacitive, pseudocapacitive and battery-type electrode materials for ESDs.29–40 In addition to the conventional considerations for ESDs (e.g., high capacitance, high power density, and long lifetime), flexibility and areal capacitance are more important for flexible ESDs. Recently, a number of MOF-related materials have been fabricated as flexible electrodes for ESDs; these materials are normally coated onto or grown in situ on flexible substrates such as plastics, textiles or carbon cloths.36,41,42 However, the challenge of developing a MOF-related electrode with good flexibility and high areal capacitance for the fabrication of flexible ESDs remains.
Cellulose is the main component of plants and the most naturally abundant biopolymer on earth. Its characteristics of intrinsic flexibility, high mechanical strength, rich organic functional groups, and sustainability make cellulose a promising substrate for the development of novel electrodes.43,44 More importantly, cellulose can also act as an intrinsic electrolyte reservoir due to its hydrophilic features and ability to swell in water.45 The hierarchical porous structure formed by crosslinking of cellulose fibers can be used to transfer the electrolyte and diffuse ions in the electrolyte to electrochemically active materials. Because of the versatile functionalities and superior properties of MOFs and cellulose, fabrication of hybrid materials based on MOFs and cellulose could combine the advantages of both materials and allow the design of novel functional materials. However, very few examples of such hybrid materials have been reported to date and the study of their applications has historically been focused only on separations (e.g. gas separation and removal of heavy metal ions and dye molecules from polluted aqueous solutions).46–49 We envision loading MOFs or MOF-related nanocrystals into a flexible cellulose substrate as a feasible approach for the design of novel flexible electrodes with promising electrochemical performance characteristics.
In this paper, we introduce an assembly approach for interweaving nanocellulose fibers, multi-walled carbon nanotubes (CNTs), and ZIF-67 (a cobalt-based zeolitic imidazolate framework, i.e. a MOF) nanocrystals into a flexible sheet. A subsequent chemical etching process converts the loaded MOF nanocrystals into Co–Ni layered double hydroxide (LDH) nanocages with unique hollow structures, a well-known electrochemically active material.50–52 This strategy involves the formation of a flexible, conductive substrate and the subsequent interweaving of the isolated LDH nanocages with nanocellulose and CNTs. The nanocellulose enables the knitting process and reinforces the LDH nanocages on the substrate, while also providing the flexibility of the composite, and the CNTs act as conductive bridges for electron transfer between the LDH nanocages. The resultant flexible and conductive composite containing compactly interconnected and homogeneously dispersed nanostructures can be used as a high performance electrode for the assembly of flexible and foldable ESDs.
Results and discussion
Cladophora cellulose (CC), a type of nanofibrillated cellulose extracted from an environmentally polluting green alga, was chosen as the substrate in this study. Firstly, it was expected that MOF nanocrystals could be loaded into its nanofibrous and mesoporous structure.53,54 Moreover, its high crystallinity55 gives rise to the outstanding mechanical flexibility of the substrate, which is an important characteristic of flexible electronic devices. Scheme 1 illustrates the synthesis procedures. Firstly, the as-synthesized ZIF-67 nanocrystals (pre-dispersed in ethanol), CC, and CNTs, with a mass ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3, are dispersed in water by sonication to form a homogeneous mixture. The dispersion is then vacuum filtered using a membrane filter, and the filter cake is dried to form a freestanding composite, called a ZIF–CC–CNT nanosheet. The composite is then immersed in an ethanol solution of nickel nitrate for three hours to etch the ZIF-67 nanocrystals. During this time, the protons generated from the hydrolysis of Ni2+ ions gradually digest the core structure of the ZIF-67 nanocrystals and release Co2+ ions into the ethanol solution. The released Co2+ ions are partly oxidized to Co3+ by the NO3− ions in the presence of the protons, followed by further co-precipitation of Ni2+ and Co2+/Co3+ and the formation of Ni–Co LDH shells, templated by ZIF-67 nanocrystals.56 Finally, a freestanding, flexible, and foldable composite, called an LDH–CC–CNT nanosheet, is obtained.
:1:3, are dispersed in water by sonication to form a homogeneous mixture. The dispersion is then vacuum filtered using a membrane filter, and the filter cake is dried to form a freestanding composite, called a ZIF–CC–CNT nanosheet. The composite is then immersed in an ethanol solution of nickel nitrate for three hours to etch the ZIF-67 nanocrystals. During this time, the protons generated from the hydrolysis of Ni2+ ions gradually digest the core structure of the ZIF-67 nanocrystals and release Co2+ ions into the ethanol solution. The released Co2+ ions are partly oxidized to Co3+ by the NO3− ions in the presence of the protons, followed by further co-precipitation of Ni2+ and Co2+/Co3+ and the formation of Ni–Co LDH shells, templated by ZIF-67 nanocrystals.56 Finally, a freestanding, flexible, and foldable composite, called an LDH–CC–CNT nanosheet, is obtained.
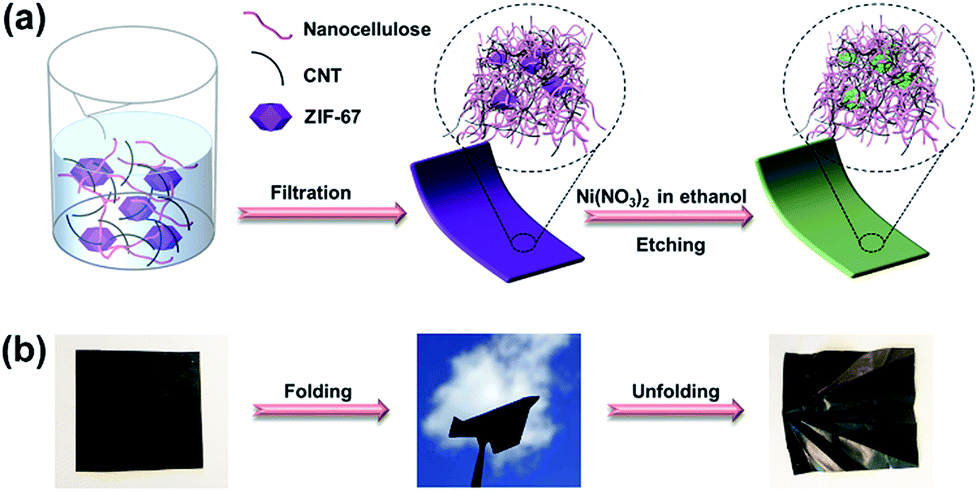 | ||
| Scheme 1 (a) Schematic illustration of the formation process of LDH–CC–CNT nanosheets; (b) optical images of a flexible, foldable LDH–CC–CNT nanosheet (size: 4 cm × 4 cm). CC = Cladophora cellulose; LDH = Co–Ni layered double hydroxide nanocages; CNTs = carbon nanotubes. | ||
With the fixed ratio of the three components in the initial dispersions, the thickness and areal density of the active materials in the hybrid nanosheets can be precisely controlled by adjusting the total amounts of the starting materials. Three ZIF–CC–CNT nanosheets and the three corresponding LDH–CC–CNT nanosheets with tuned thicknesses (9, 39, and 103 μm) were prepared. Thermogravimetric analysis (Fig. S1†) indicated that the content of the active material ZIF-67 was 0.305, 0.754, and 1.720 mg cm−2 in the ZIF–CC–CNT nanosheet and LDH was 0.225, 0.481, and 1.452 mg cm−2 in the LDH–CC–CNT nanosheet.
The structures of the ZIF–CC–CNT and LDH–CC–CNT nanosheets were fully characterized by powder X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and X-ray energy dispersive spectroscopy (EDS). The XRD pattern of ZIF–CC–CNT in Fig. 1a clearly shows the existence of ZIF-67, cellulose and CNTs, thus confirming the composition of the nanosheet. The diffraction peaks for ZIF-67 were entirely absent after etching the composite in an ethanol solution of Ni(NO3)2·6H2O, and the peaks for a typical LDH structure were found, confirming the formation of an LDH structure by the reaction between ZIF-67 and Ni(NO3)2·6H2O (Fig. 1a and S2†). Strong and narrow diffraction peaks at 2θ = 14, 17 and 22° in both the ZIF–CC–CNT and the LDH–CC–CNT nanosheets confirmed the high crystallinity of the CC nanofibers.57 The elemental composition of LDH–CC–CNT was confirmed by EDS and XPS studies. The EDS spectrum of the selected area of LDH–CC–CNT (Fig. S3†) revealed that the LDH nanocages contained Co and Ni, with a Co:Ni ratio of 1:2.1. The XPS survey scan spectrum of LDH–CC–CNT (Fig. S4†) indicated the presence of C, O, N, Ni, and Co in the composite. Fig. 1c and d show the high resolution XPS spectra of LDH–CC–CNT in the Co 2p3/2 and Ni 2p3/2 regions. The deconvoluted peaks at 782.3 and 780.8 eV in the Co 2p3/2 spectrum correspond to Co2+ and Co3+, respectively,58 confirming that a portion of the Co2+ ions released from ZIF-67 were oxidized to Co3+ during the etching procedure. The single peak located at 856.1 eV in the Ni 2p3/2 spectrum indicated the existence of Ni2+ in the LDH structure.50
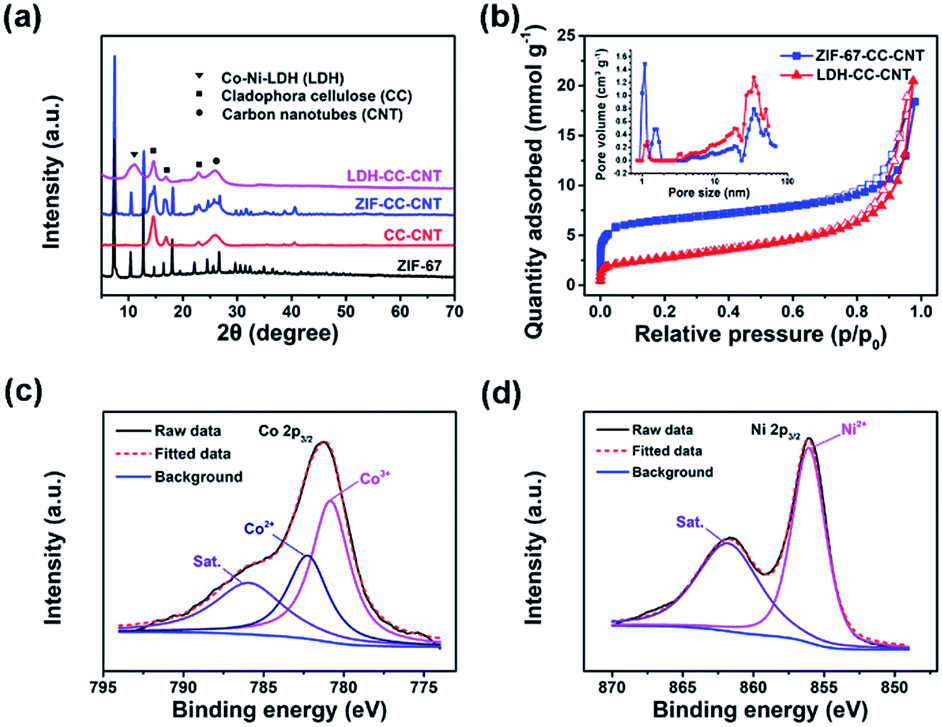 | ||
| Fig. 1 (a) A comparison of powder X-ray diffraction patterns of pure ZIF-67, Cladophora cellulose (CC)–CNTs, ZIF–CC–CNT nanosheets, and LDH–CC–CNT nanosheets; (b) N2 adsorption and desorption isotherms of ZIF–CC–CNT and LDH–CC–CNT nanosheets, recorded at 77 K. The inset shows the pore size distributions calculated from the adsorption branches using density functional theory; (c) high-resolution Co 2p3/2 and (d) Ni 2p3/2 X-ray photoelectron spectra of LDH–CC–CNT. CNT = carbon nanotube; LDH = Co–Ni layered double hydroxide nanocages. | ||
Nitrogen sorption measurements at 77 K were carried out to analyze the porosity of the samples. As shown in Fig. 1b, the adsorption isotherm for ZIF–CC–CNT, with rapid N2 uptake at low partial pressures (P/P0 < 0.1), revealed the microporous structure of ZIF-67. Meanwhile, the pore size distribution analysis based on the adsorption isotherm indicated that ZIF–CC–CNT had relatively narrow micropores, with pore sizes centered at 1.0 and 1.4 nm, whereas these typical micropores were absent in LDH–CC–CNT because of the digestion of the framework of ZIF-67 during the etching procedure. Accordingly, the calculated Brunauer–Emmett–Teller (BET) surface areas for LDH–CC–CNT of different thicknesses lie in the range of 219 to 261 m2 g−1; these were significantly smaller than those for ZIF–CC–CNT (525–693 m2 g−1). The size of the micropores in LDH–CC–CNT (1.2 nm) can be attributed to the presence of CNTs (Fig. S5†). Both ZIF–CC–CNT and LDH–CC–CNT samples showed mesoporous structures generated by interweaving CC nanofibers and CNTs with pore size distributions in the range of 20.0 to 34.4 nm.
The morphology of the ZIF–CC–CNT and LDH–CC–CNT nanosheets was studied by field emission scanning electron microscopy (FESEM) to reveal the interconnections between the ZIF-67 or LDH nanocrystals, the CC nanofibers, and the CNTs. The as-synthesized ZIF-67 nanocrystals had a typical rhombic dodecahedral morphology and an average size of approximately 800 nm (Fig. S6a†). SEM images (Fig. 2) revealed the surface morphology of the ZIF–CC–CNT and LDH–CC–CNT nanosheets at different magnifications. The cross-linked CC nanofibers and CNTs (with diameters of about 30 nm and 10 nm, respectively) were interwoven in the well-dispersed ZIF-67 crystals to form the framework of the ZIF–CC–CNT nanosheets. More importantly, the ZIF-67 nanocrystals were not only attached to the surface of the nanosheets but were also entangled in the matrix, as indicated by the “bumps” in the SEM images, suggesting that ZIF-67 nanocrystals were evenly distributed throughout the entire nanosheet. In comparison, in the ZIF–CNT composite, the ZIF-67 nanocrystals formed agglomerates with the CNTs, which remained separate in the absence of cellulose nanofibers (Fig. S6b and c†). Therefore, we can conclude that CC nanofibers play a key role in the formation of uniform and intimately connected nanostructures because of their ability to aid in the dispersion and intertwining of the active materials.
 | ||
| Fig. 2 Scanning electron microscopy images of (a–c) ZIF–CC–CNT and (d–f) LDH–CC–CNT nanosheets at different magnifications. CC = Cladophora cellulose; CNT = carbon nanotube; LDH = Co–Ni layered double hydroxide nanocages. | ||
After etching the ZIF–CC–CNT nanosheets in a solution of Ni(NO3)2·6H2O in ethanol, where the ZIF-67 nanocrystals were transformed into hollow LDH nanocages, these uniform and interconnected nanostructures were successfully transformed into LDH–CC–CNT nanosheets. It was expected that the uniformity of the LDH–CC–CNT nanosheets and the hollow nanostructure of LDH would lead to fully accessible sites for the active materials and fast diffusion of guest molecules or ions. Furthermore, the intimate connections between the LDH nanocages and the flexible and conductive matrix composed of nanocellulose and CNTs could effectively improve the conductivity of LDH. Considering the flexible, binder-free, conductive, and fully accessible characteristics of these structures, we anticipated that LDH–CC–CNT nanosheets would make effective electrode materials.
The electrochemical performance of the LDH–CC–CNT electrodes was studied in a traditional three-electrode cell using 1.0 M aqueous KOH as the electrolyte. Binder-free electrodes were prepared by pressing a piece of LDH–CC–CNT nanosheet (1 × 2 cm) at a pressure of 150 MPa onto a piece of flexible graphite paper as the current collector. The design of these binder-free electrodes was chosen to avoid “dead” mass and volume in the electrode, thus increasing its gravimetric/volumetric capacity. Fig. 3a shows the cyclic voltammetry (CV) curves of the LDH–CC–CNT (9 μm, 0.225 mg cm−2 LDH) electrode at different scan rates (1–30 mV s−1) in the potential range of 0 to 0.6 V vs. Hg/HgO. With the increasing scan rate from 1 to 30 mV, the CV curves were similar in shape, with slightly shifted peak positions. The CV curve at a scan rate of 1 mV s−1 had a distinct pair of redox peaks at 0.40 and 0.29 V vs. Hg/HgO, related to the electrochemical redox reactions of Ni(OH)2/NiOOH and CoOOH/CoO2 in LDH.50 It should be noted that it is difficult to distinguish redox peaks for these two reactions because of the similar redox potentials for Ni2+/Ni3+ and Co3+/Co4+.56 In addition, a pair of broad and inconspicuous redox peaks in the potential range of 0.1 to 0.2 V vs. Hg/HgO was observed in the CV curve. This pair of redox peaks at low potentials can be ascribed to the redox reaction of Co(OH)2/CoOOH and further confirms the existence of Co2+ in the LDH structure.59 Based on this analysis of the CV curves, we hypothesized that the electrochemical reaction process in the LDH is as follows:
| Co(OH)2 + OH− ↔ CoOOH + H2O + e− | (1) |
| CoOOH + OH− ↔ CoO2 + H2O + e− | (2) |
| Ni(OH)2 + OH− ↔ NiOOH + H2O + e− | (3) |
| ip = aνb | (4) |
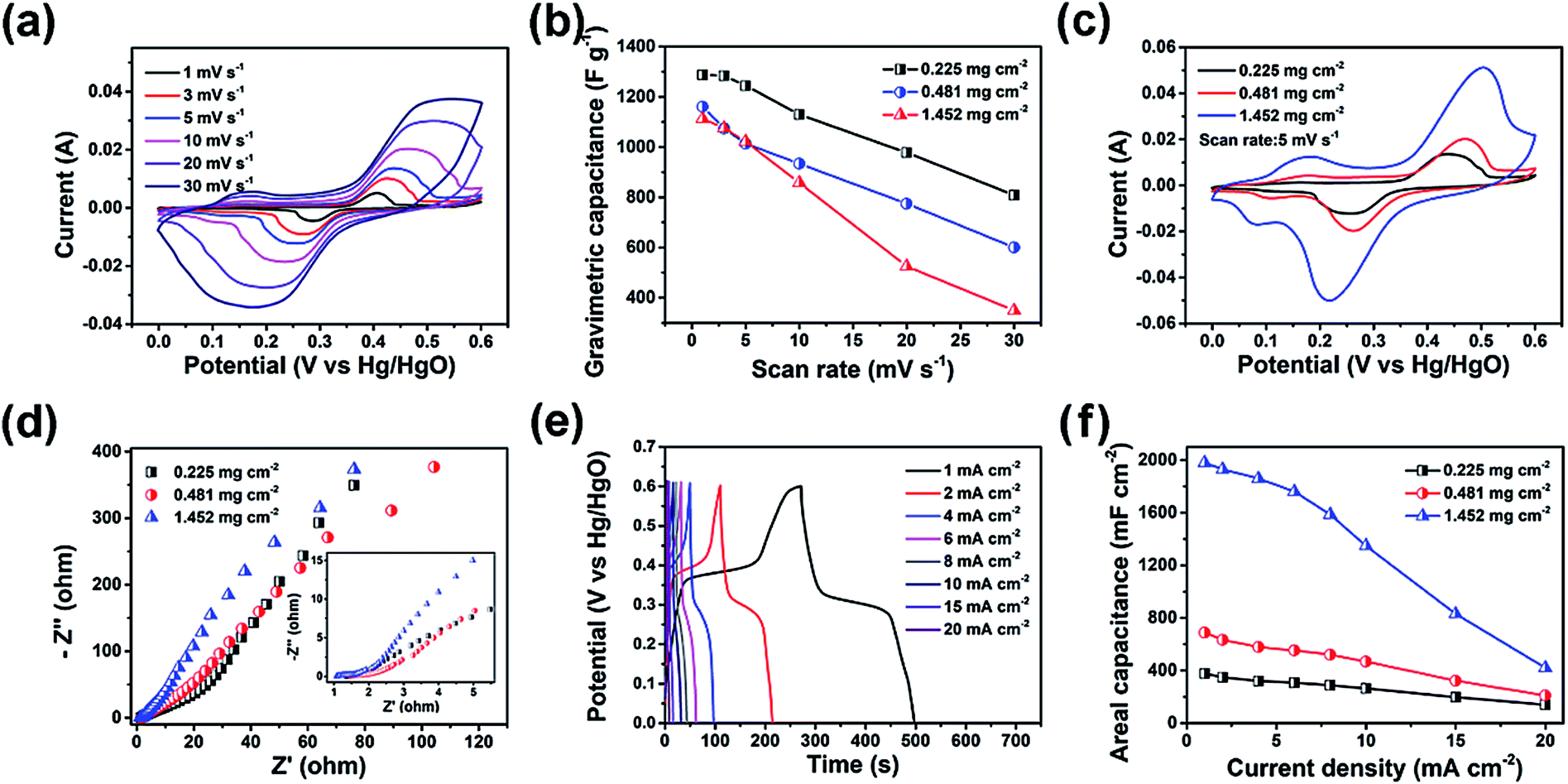 | ||
| Fig. 3 (a) Cyclic voltammetry (CV) curves of LDH–CC–CNT with a mass loading of 0.225 mg cm−2 LDH at different scan rates. (b) Gravimetric capacitance and rate performance of LDH in the LDH–CC–CNT electrodes with different areal densities (values on the vertical axis are normalized with respect to the LDH content of the electrode). (c) CV curves of LDH–CC–CNT with different mass loadings of LDH at a scan rate of 5 mV s−1. (d) Nyquist plots of LDH–CC–CNT electrodes; the inset shows a close-up of the high frequency data. (e) Galvanostatic charge–discharge (GCD) curves of an LDH–CC–CNT electrode (0.225 mg cm−2 LDH). (f) Areal capacitance and rate performance of LDH–CC–CNT electrodes with different areal densities. CC = Cladophora cellulose; CNT = carbon nanotube; LDH = Co–Ni layered double hydroxide nanocages. | ||
Generally, the peak current is proportional to the square root of the scan rate (b = 0.5) for a battery-type faradaic reaction, whereas the pseudocapacitive current is linearly dependent on the scan rate (b = 1). We found that the plots of peak current vs. scan rate can be linearly fitted with b = 0.5 for the three electrodes of LDH–CC–CNT with different loadings of LDH (Fig. S7†), suggesting that the LDH–CC–CNT electrodes are battery-type electrodes and the charge storage mechanism is mainly based on the diffusion-controlled intercalation/deintercalation of the electrolyte ions.59–61
The capacitances of the active material (LDH) and the entire electrode were calculated by analyzing the CV curves. In order to evaluate the contributions of the CNTs and the current collector to the capacitance of the electrode, a background electrode composed of CC and CNTs on graphite paper was prepared for electrochemical analysis. Based on the CV curves of LDH–CC–CNT and this CC–CNT electrode at 1 mV s−1 and by deducting the contributions of the CNTs and graphite paper, the specific capacitance of LDH was calculated to be 1287.8 F g−1, which is comparable to the reported values for Co–Ni LDH.50,51,62–64 Importantly, the gravimetric capacitance of LDH was not significantly reduced in the electrode with a higher mass loading, as shown in Fig. 3b. In contrast, the capacitance of LDH was only 732.2 F g−1 in an LDH–PVDF–CNT electrode prepared by coating LDH, CNTs and a binder of polyvinylidene fluoride (PVDF) on graphite paper in a traditional manner, under the same conditions (Fig. S8†). Comparison of the nanostructures in the SEM images provides a possible explanation for this large difference in the capacitive performance of LDH in the two electrodes. As mentioned above, the LDH nanocages were uniformly dispersed and interconnected by cellulose nanofibers and CNTs in the LDH–CC–CNT nanosheets. In contrast, the LDH–PVDF–CNT electrode displayed agglomerates of LDH nanocages attached onto the surface of the CNT matrix (Fig. S6d and e†). Consequently, the rates of ion diffusion and electron transfer were limited, leading to the much lower capacitance. These results strongly supported our assumption that the electrochemical performance of the electrode could be significantly increased by forming a uniform and interwoven nanostructure within the conductive substrate.
For flexible and wearable electronic devices, areal capacitance is even more important than gravimetric capacitance. In general, the areal capacitance of an electrode increases with the increasing areal density of the active material in the substrate. However, aggregation and stacking of the active materials usually occur with subsequently increased resistance.65 As a result, the areal capacitance will gradually reach a plateau when the mass loading is increased, and the electrochemical performance of the active material will be much lower than the theoretical value. In order to evaluate the effects of mass loading and thickness on the electrochemical performance, LDH–CC–CNT electrodes with different mass loadings (0.225, 0.481, and 1.452 mg cm−2 LDH) and thicknesses (9, 39, and 103 μm) were studied using CV (Fig. S9†). Fig. 3c shows a comparison of the CV curves of the three electrodes at the same scan rate of 5 mV s−1. Notably, the redox peak positions only shifted slightly with the increasing thickness and mass loading. Meanwhile, the electrochemical impedance spectroscopy (EIS) results displayed as Nyquist plots in Fig. 3d revealed that both the high frequency resistance and the charge transfer resistance for LDH–CC–CNT electrodes were relatively low, and these resistances were not significantly affected by increasing the mass loading and thickness because of the uniform nanostructure of the conductive electrodes.
Galvanostatic charge–discharge (GCD) measurements were performed to evaluate the energy storage performance of the LDH–CC–CNT electrodes (Fig. S10†). The highly symmetrical shape of the chronopotentiometric curves reflected the reversible redox reactions in the electrode (Fig. 3e). The discharge curves showed that the areal capacitances of the LDH–CC–CNT electrodes with mass loadings of 0.225, 0.481, and 1.452 mg cm−2 LDH were 377.5, 688.6 and 1979.2 mF cm−2, respectively, at a current density of 1 mA cm−2. The three electrodes retained 37.1, 30.5, and 21.2% of their capacitances when the current density was increased from 1 to 20 mA cm−2, indicating a good rate capability (Fig. 3f). Importantly, the flexibility of the electrode was not influenced by increasing the thickness and mass loading towards higher areal capacitances. A further increase in the mass loading of LDH to 2.87 mg cm−2 in the LDH–CC–CNT nanosheet with a thickness of 212 μm achieved an even higher areal capacitance of 3200 mF cm−2; however, the electrode had a very poor rate capability and was not as flexible. Nonetheless, the value of 1979.2 mF cm−2 for the areal capacitance of the LDH–CC–CNT electrode ranks as one of the highest reported for LDH-based electrodes (Co–Ni LDH/NiCo2O4/carbon fiber paper:66 1640 mF cm−2 at 2 mA cm−2; NiCo2S4@Ni–Mn LDH/graphene sponge:67 1740 mF cm−2 at 1 mA cm−2; Ni–Co@Ni–Co LDH nanotube array/carbon fiber cloth:62 2000 mF cm−2 at 4.6 mA cm−2; NiCo-LDH/carbon fiber cloth:59 2242 mF cm−2 at 1 mA cm−2).
To demonstrate the potential of the composite electrode for practical applications in flexible electronics, we assembled a flexible, foldable, and all-solid-state hybrid ESD. As shown in Fig. 4a, the LDH–CC–CNT (1 cm × 2 cm, 0.481 mg cm−2 LDH) and CC–CNT (1 cm × 2 cm, 3.375 mg cm−2 CNTs) nanosheets were used as the positive and negative electrodes, respectively. A poly(vinyl alcohol)/KOH gel was used as the solid electrolyte, and a piece of filter paper (1.2 cm × 2.5 cm) was used as the separator. The total volume of the assembled device was 0.2 cm3. The CV curves in Fig. 4b present the potential windows of −1.0 to 0 V vs. Hg/HgO for the LDH–CC–CNT electrode and 0 to 0.6 V vs. Hg/HgO for the CC–CNT electrode at the same scan rate of 5 mV s−1. It was anticipated that this hybrid ESD could reach a high operating potential. CV curves of the hybrid ESD were recorded in the two-electrode system at a scan rate of 50 mV s−1 within the potential window from 0–1 V to 0–1.6 V (vs. Hg/HgO), as shown in Fig. 4c. This demonstrated that the stable operating voltage could be extended to 1.6 V. In addition, CV curves of the hybrid ESD at different scan rates (10–50 mV s−1) were measured within the potential window of 0–1.6 V vs. Hg/HgO (Fig. S11†). The almost rectangular shapes of the CV curves at low operating potentials demonstrated the double-layer capacitance of CC–CNT. Broad redox peaks were observed at high operating potentials, indicating a combined contribution of both capacity and double-layer capacitance in the hybrid ESD. To evaluate the flexibility and foldability of the hybrid ESD, we recorded the CV curves when the device was folded to 90° and 180°. As shown in Fig. 4d, the CV curves were only very slightly different before and during folding, indicating that the device was fully flexible and foldable.
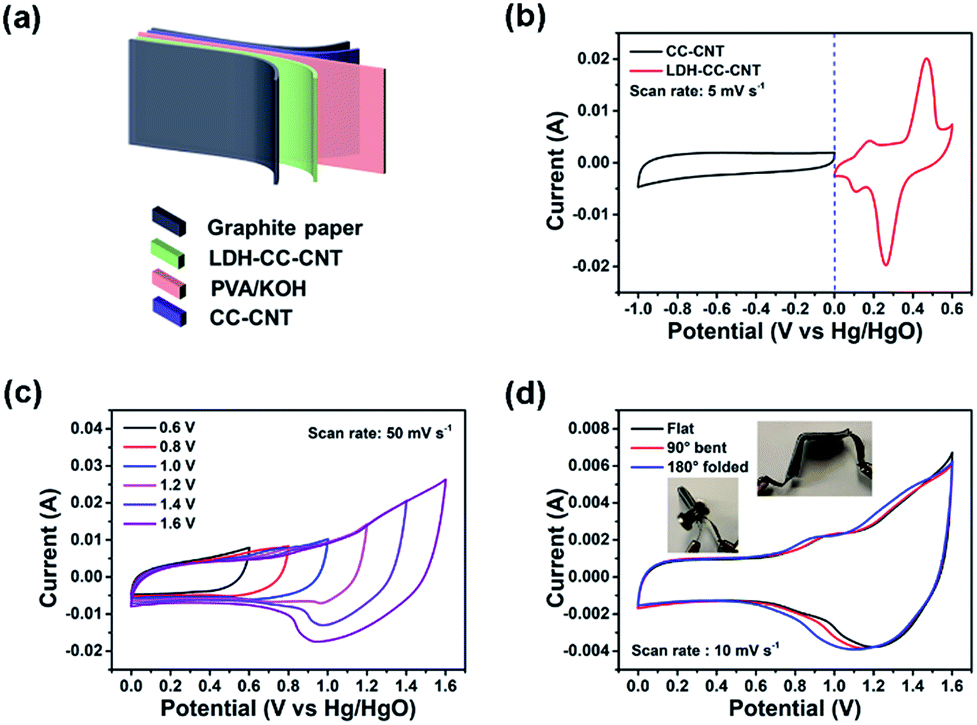 | ||
| Fig. 4 (a) Schematic illustration of the flexible and solid-state hybrid ESD LDH–CC–CNT//CC–CNT. (b) Cyclic voltammetry (CV) curves of LDH–CC–CNT and CC–CNT electrodes at the same scan rate. (c) CV curves of the device at different potential windows at the same scan rate. (d) CV curves of the device under different bending conditions. ESD = energy storage device; CC = Cladophora cellulose; CNT = carbon nanotube; LDH = Co–Ni layered double hydroxide. | ||
The specific capacitance, and energy and power density of the hybrid ESD were calculated from the GCD measurements. The areal capacitance for the whole cell was 168.3, 149.5, 123.8, and 102.5 mF cm−2 at discharge current densities of 1, 4, 10, and 20 mA cm−2, respectively (Fig. 5a). Accordingly, the maximum energy and power densities of the hybrid ESD were 0.6 mW h cm−3 (60 μW h cm−2) and 160 mW cm−3 (16 mW cm−2), respectively (Fig. 5b). The values of areal capacitance, and energy and power density were comparable to those for state-of-the-art flexible SCs (Table S1†).29,67–71 It should be noted that the volume of the whole hybrid ESD was considered when calculating the volumetric energy/power densities. We also assembled two hybrid ESD units in series to power an LED. As seen in Fig. 5c, the devices in the series light the LED even when they were folded. The long-term stability of the hybrid ESD was tested through continuous CV cycling. As shown in Fig. 5d, the hybrid device retained more than 80% of its original capacitance after 2000 cycles at 50 mV s−1. Collectively, the electrochemical studies suggested that the all-solid-state hybrid ESD showed an excellent capacitive behavior, a good rate capability, and excellent flexibility and foldability.
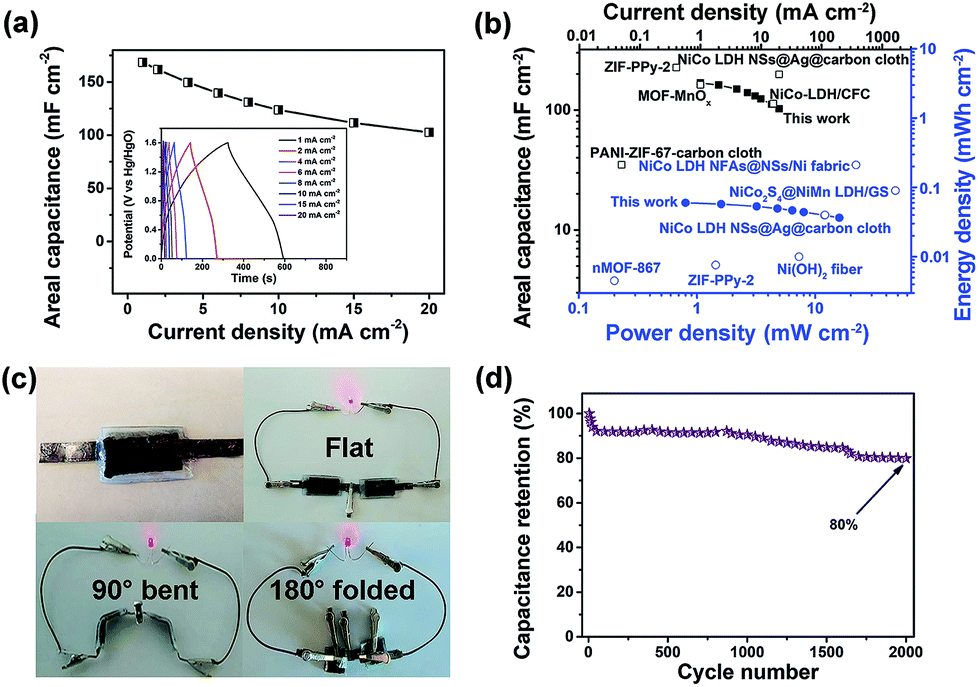 | ||
| Fig. 5 (a) Galvanostatic charge–discharge curves of the flexible and solid-state hybrid ESD LDH–CC–CNT//CC–CNT and the calculated areal capacitances at different current densities. (b) Comparison of areal capacitance (■ this work, □ reported values), and power and energy density (● this work, ○ reported values) of the device with those of other flexible ESDs based on electrodes of nMOF-867,29 MOF–MnOx,41 PANI-ZIF-67-carbon cloth,42 NiCo-LDH/CFC,59 NiCo2S4@NiMn-LDH/GS,67 NiCo-LDH NSs@Ag@carbon cloth,68 NiCo-LDH NFAs@NSs/Ni fabric,69 ZIF-PPy-2,70 Ni(OH)2 nanofibers71 and various carbon electrodes. (c) Photo of red LEDs powered by the bent or folded device. (d) Cycling performance of the device. ESD = energy storage device; CC = Cladophora cellulose; CNT = carbon nanotube; LDH = layered double hydroxide; PANI = polyaniline; PPy = polypyrrole; CFC = carbon fiber cloth; NFAs = nanoflake arrays; NSs = nanosheets; GS = graphene sponge. | ||
Conclusions
In conclusion, novel LDH–CC–CNT nanosheets have been successfully prepared by interweaving MOF-derived Co–Ni LDH nanocages with CC nanofibers and CNTs. The electrodes based on LDH–CC–CNT nanosheets had a very high areal capacitance and a good rate capability. Given their excellent flexibility and foldability, along with their superior electrochemical performance, the LDH–CC–CNT electrodes were fabricated into an all-solid-state hybrid ESD. We conclude that the interwoven, hierarchically porous, fully accessible, and conductive nanostructures in the LDH–CC–CNT nanosheets play a key role in the electrochemical performance of the hybrid ESD. An external current collector is required for this electrode and energy storage device because of the limited interactions between the hydrophobic CNTs, and the hydrophilic nanocellulose and LDH nanocages. This limitation could be overcome by using chemically modified nanocellulose and CNTs in high quality dispersions to design highly compact and conductive matrices.72 Thus, this study opens up a path for the development of new designs for thin, flexible, lightweight, and high-performance electronic energy storage devices based on MOF-related materials and sustainable cellulose.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was supported by an Åforsk research grant (18-413), a STINT initiation grant (IB2017-6985), the National Natural Science Foundation of China (21504043), and the Natural Science Foundation of Jiangsu Province (BK20170994). We thank Prof. Martin Sjödin and Dr Zhaohui Wang for valuable discussions.Notes and references
- V. L. Pushparaj, M. M. Shaijumon, A. Kumar, S. Murugesan, L. Ci, R. Vajtai, R. J. Linhardt, O. Nalamasu and P. M. Ajayan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13574–13577 CrossRef CAS PubMed.
- L. Hu and Y. Cui, Energy Environ. Sci., 2012, 5, 6423–6435 RSC.
- X. Wang, X. Lu, B. Liu, D. Chen, Y. Tong and G. Shen, Adv. Mater., 2014, 26, 4763–4782 CrossRef CAS PubMed.
- H. Wang, Y. Yang and L. Guo, Adv. Energy Mater., 2016, 7, 1601709 CrossRef.
- G. Nyström, A. Razaq, M. Strømme, L. Nyholm and A. Mihranyan, Nano Lett., 2009, 9, 3635–3639 CrossRef PubMed.
- Z. Wang, P. Tammela, M. Strømme and L. Nyholm, Adv. Energy Mater., 2017, 7, 1700130 CrossRef.
- Q. Meng, K. Cai, Y. Chen and L. Chen, Nano Energy, 2017, 36, 268–285 CrossRef CAS.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- J. H. Park and O. O. Park, J. Power Sources, 2002, 109, 121–126 CrossRef CAS.
- V. Subramanian, H. Zhu and B. Wei, J. Power Sources, 2006, 159, 361–364 CrossRef CAS.
- T. Liu, L. Finn, M. Yu, H. Wang, T. Zhai, X. Lu, Y. Tong and Y. Li, Nano Lett., 2014, 14, 2522–2527 CrossRef CAS PubMed.
- Y. Shao, M. F. El-Kady, J. Sun, Y. Li, Q. Zhang, M. Zhu, H. Wang, B. Dunn and R. B. Kaner, Chem. Rev., 2018, 118, 9233–9280 CrossRef CAS PubMed.
- N. Choudhary, C. Li, J. Moore, N. Nagaiah, L. Zhai, Y. Jung and J. Thomas, Adv. Mater., 2017, 29, 1605336 CrossRef PubMed.
- T. Brousse, M. Toupin and D. Bélanger, J. Electrochem. Soc., 2004, 151, A614–A622 CrossRef CAS.
- J. Huang, P. Xu, D. Cao, X. Zhou, S. Yang, Y. Li and G. Wang, J. Power Sources, 2014, 246, 371–376 CrossRef CAS.
- J. Bae, K. Song Min, J. Park Young, M. Kim Jong, M. Liu and Z. L. Wang, Angew. Chem., Int. Ed., 2011, 50, 1683–1687 CrossRef CAS PubMed.
- M. F. El-Kady and R. B. Kaner, Nat. Commun., 2013, 4, 1475 CrossRef PubMed.
- L. Liu, Z. Niu, L. Zhang, W. Zhou, X. Chen and S. Xie, Adv. Mater., 2014, 26, 4855–4862 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabrés i Xamena and J. Gascon, Nat. Mater., 2015, 14, 48–55 CrossRef CAS PubMed.
- W. Zhang, B. Zheng, W. Shi, X. Chen, Z. Xu, S. Li, R. Chi Yonggui, Y. Yang, J. Lu, W. Huang and F. Huo, Adv. Mater., 2018, 30, 1800643 CrossRef PubMed.
- Y. Chen, P. Li, J. A. Modica, R. J. Drout and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 5678–5681 CrossRef CAS PubMed.
- G. Lu and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 7832–7833 CrossRef CAS PubMed.
- L. Wang, Y. Han, X. Feng, J. Zhou, P. Qi and B. Wang, Coord. Chem. Rev., 2016, 307, 361–381 CrossRef CAS.
- X. Cao, C. Tan, M. Sindoro and H. Zhang, Chem. Soc. Rev., 2017, 46, 2660–2677 RSC.
- H. B. Wu and X. W. Lou, Sci. Adv., 2017, 3, eaap9252 CrossRef PubMed.
- K. M. Choi, H. M. Jeong, J. H. Park, Y.-B. Zhang, J. K. Kang and O. M. Yaghi, ACS Nano, 2014, 8, 7451–7457 CrossRef CAS PubMed.
- J. Yang, P. Xiong, C. Zheng, H. Qiu and M. Wei, J. Mater. Chem. A, 2014, 2, 16640–16644 RSC.
- W. H. Li, K. Ding, H. R. Tian, M. S. Yao, B. Nath, W. H. Deng, Y. Wang and G. Xu, Adv. Funct. Mater., 2017, 27, 1702067 CrossRef.
- D. Sheberla, J. C. Bachman, J. S. Elias, C.-J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2016, 16, 220–224 CrossRef PubMed.
- D. Feng, T. Lei, M. R. Lukatskaya, J. Park, Z. Huang, M. Lee, L. Shaw, S. Chen, A. A. Yakovenko, A. Kulkarni, J. Xiao, K. Fredrickson, J. B. Tok, X. Zou, Y. Cui and Z. Bao, Nat. Energy, 2018, 3, 30–36 CrossRef CAS.
- F. Meng, Z. Fang, Z. Li, W. Xu, M. Wang, Y. Liu, J. Zhang, W. Wang, D. Zhao and X. Guo, J. Mater. Chem. A, 2013, 1, 7235–7241 RSC.
- R. Bendi, V. Kumar, V. Bhavanasi, K. Parida and S. Lee Pooi, Adv. Energy Mater., 2015, 6, 1501833 CrossRef.
- C. Guan, X. Liu, W. Ren, X. Li, C. Cheng and J. Wang, Adv. Energy Mater., 2017, 7, 1602391 CrossRef.
- P. Pachfule, D. Shinde, M. Majumder and Q. Xu, Nat. Chem., 2016, 8, 718–724 CrossRef CAS PubMed.
- R. R. Salunkhe, Y. V. Kaneti, J. Kim, J. H. Kim and Y. Yamauchi, Acc. Chem. Res., 2016, 49, 2796–2806 CrossRef CAS PubMed.
- Q. Liu, X. Liu, C. Shi, Y. Zhang, X. Feng, M.-L. Cheng, S. Su and J. Gu, Dalton Trans., 2015, 44, 19175–19184 RSC.
- X. Liu, C. Shi, C. Zhai, M. Cheng, Q. Liu and G. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 4585–4591 CrossRef CAS PubMed.
- Y.-Z. Zhang, T. Cheng, Y. Wang, W.-Y. Lai, H. Pang and W. Huang, Adv. Mater., 2016, 28, 5242–5248 CrossRef CAS PubMed.
- L. Wang, X. Feng, L. Ren, Q. Piao, J. Zhong, Y. Wang, H. Li, Y. Chen and B. Wang, J. Am. Chem. Soc., 2015, 137, 4920–4923 CrossRef CAS PubMed.
- L. Nyholm, G. Nyström, A. Mihranyan and M. Strømme, Adv. Mater., 2011, 23, 3751–3769 CAS.
- M. Hamedi, E. Karabulut, A. Marais, A. Herland, G. Nyström and L. Wågberg, Angew. Chem., Int. Ed., 2013, 52, 12038–12042 CrossRef CAS PubMed.
- Z. Gui, H. Zhu, E. Gillette, X. Han, G. W. Rubloff, L. Hu and S. B. Lee, ACS Nano, 2013, 7, 6037–6046 CrossRef CAS PubMed.
- M. Matsumoto and T. Kitaoka, Adv. Mater., 2016, 28, 1765–1769 CrossRef CAS PubMed.
- H. Zhu, X. Yang, E. D. Cranston and S. Zhu, Adv. Mater., 2016, 28, 7652–7657 CrossRef CAS PubMed.
- J. Park and M. Oh, Nanoscale, 2017, 9, 12850–12854 RSC.
- L. Zhu, L. Zong, X. Wu, M. Li, H. Wang, J. You and C. Li, ACS Nano, 2018, 12, 4462–4468 CrossRef CAS PubMed.
- Z. Jiang, Z. Li, Z. Qin, H. Sun, X. Jiao and D. Chen, Nanoscale, 2013, 5, 11770–11775 RSC.
- G. Nagaraju, G. S. R. Raju, Y. H. Ko and J. S. Yu, Nanoscale, 2016, 8, 812–825 RSC.
- X. Li, D. Du, Y. Zhang, W. Xing, Q. Xue and Z. Yan, J. Mater. Chem. A, 2017, 5, 15460–15485 RSC.
- H. Olsson, D. O. Carlsson, G. Nyström, M. Sjödin, L. Nyholm and M. Strømme, J. Mater. Sci., 2012, 47, 5317–5325 CrossRef CAS.
- R. Pan, O. Cheung, Z. Wang, P. Tammela, J. Huo, J. Lindh, K. Edström, M. Strømme and L. Nyholm, J. Power Sources, 2016, 321, 185–192 CrossRef CAS.
- A. Mihranyan, A. P. Llagostera, R. Karmhag, M. Strømme and R. Ek, Int. J. Pharm., 2004, 269, 433–442 CrossRef CAS PubMed.
- H. Hu, B. Guan, B. Xia and X. W. Lou, J. Am. Chem. Soc., 2015, 137, 5590–5595 CrossRef CAS PubMed.
- A. Mihranyan, J. Appl. Polym. Sci., 2010, 119, 2449–2460 CrossRef.
- C. Zhang, J. Zhao, L. Zhou, Z. Li, M. Shao and M. Wei, J. Mater. Chem. A, 2016, 4, 11516–11523 RSC.
- T. Wang, S. Zhang, X. Yan, M. Lyu, L. Wang, J. Bell and H. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 15510–15524 CrossRef CAS PubMed.
- H. Liang, J. Lin, H. Jia, S. Chen, J. Qi, J. Cao, T. Lin, W. Fei and J. Feng, J. Mater. Chem. A, 2018, 6, 15040–15046 RSC.
- T. Brousse, D. Bélanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- Y. Liu, N. Fu, G. Zhang, M. Xu, W. Lu, L. Zhou and H. Huang, Adv. Funct. Mater., 2017, 27, 1605307 CrossRef.
- T. Li, G. H. Li, L. H. Li, L. Liu, Y. Xu, H. Y. Ding and T. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 2562–2572 CrossRef CAS PubMed.
- H. Wu, Y. Zhang, W. Yuan, Y. Zhao, S. Luo, X. Yuan, L. Zheng and L. Cheng, J. Mater. Chem. A, 2018, 6, 16617–16626 RSC.
- Y. Ko, M. Kwon, W. K. Bae, B. Lee, S. W. Lee and J. Cho, Nat. Commun., 2017, 8, 536 CrossRef PubMed.
- L. Huang, D. Chen, Y. Ding, S. Feng, Z. L. Wang and M. Liu, Nano Lett., 2013, 13, 3135–3139 CrossRef CAS PubMed.
- H. Wan, J. Liu, Y. Ruan, L. Lv, L. Peng, X. Ji, L. Miao and J. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 15840–15847 CrossRef CAS PubMed.
- S. C. Sekhar, G. Nagaraju and J. S. Yu, Nano Energy, 2017, 36, 58–67 CrossRef CAS.
- G. Nagaraju, S. Chandra Sekhar, L. Krishna Bharat and J. S. Yu, ACS Nano, 2017, 11, 10860–10874 CrossRef CAS PubMed.
- X. Xu, J. Tang, H. Qian, S. Hou, Y. Bando, M. S. A. Hossain, L. Pan and Y. Yamauchi, ACS Appl. Mater. Interfaces, 2017, 9, 38737–38744 CrossRef CAS PubMed.
- X. Dong, Z. Guo, Y. Song, M. Hou, J. Wang, Y. Wang and Y. Xia, Adv. Funct. Mater., 2014, 24, 3405–3412 CrossRef CAS.
- A. Hajian, S. B. Lindström, T. Pettersson, M. M. Hamedi and L. Wågberg, Nano Lett., 2017, 17, 1439–1447 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta10133g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |