
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompaction of a zirconium metal–organic framework (UiO-66) for high density hydrogen storage applications†
Sonwabo E.
Bambalaza
ab,
Henrietta W.
Langmi
 *a,
Robert
Mokaya
c,
Nicholas M.
Musyoka
*a,
Jianwei
Ren
ad and
Lindiwe E.
Khotseng
b
*a,
Robert
Mokaya
c,
Nicholas M.
Musyoka
*a,
Jianwei
Ren
ad and
Lindiwe E.
Khotseng
b
aHySA Infrastructure Centre of Competence, Energy Centre, Council for Scientific and Industrial Research (CSIR), PO Box 395, Pretoria 0001, South Africa. E-mail: hlangmi@csir.co.za
bFaculty of Natural Science, University of the Western Cape, Bellville, Cape Town 7535, South Africa
cSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
dMechanical Engineering Science Department, University of Johannesburg, Johannesburg, South Africa
First published on 12th November 2018
Abstract
We report a rare case whereby a metal–organic framework (MOF), namely UiO-66, is compacted at high pressure (∼700 MPa or 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 psi) resulting in densification and improved total volumetric hydrogen storage capacity, but crucially, without compromising the total gravimetric uptake attained in the powdered form of the MOF. The applied compaction pressure is also unprecedented for MOFs as most studies have shown the MOF structure to collapse when compacted at very high pressure. The UiO-66 prepared in this study retained ∼98% of the original surface area and microporosity after compaction at ∼700 MPa, and the densified pellets achieved a total H2 uptake of 5.1 wt% at 100 bar and 77 K compared to 5.0 wt% for the UiO-66 powder. Depending on the method used to compute the volumetric uptake, the densified UiO-66 attained unprecedented volumetric capacity at 77 K and 100 bar of up to 74 g L−1 (13 g L−1 at 298 K) compared to 29 g L−1 for the powder (6 g L−1 at 298 K) using a conventional method that takes into account the packing density of the adsorbents, or 43 g L−1 (compared to 35 g L−1 for the powder at 77 K and 100 bar) based on a method that uses both the single crystal and skeletal densities of MOFs. However, regardless of the difference in the calculated values according to the two methods, the concept of UiO-66 compaction for improving volumetric capacity without compromising gravimetric uptake is clearly proven in this study and shows promise for the achievement of hydrogen storage targets for a single material as set by the United States Department of Energy (DOE).
000 psi) resulting in densification and improved total volumetric hydrogen storage capacity, but crucially, without compromising the total gravimetric uptake attained in the powdered form of the MOF. The applied compaction pressure is also unprecedented for MOFs as most studies have shown the MOF structure to collapse when compacted at very high pressure. The UiO-66 prepared in this study retained ∼98% of the original surface area and microporosity after compaction at ∼700 MPa, and the densified pellets achieved a total H2 uptake of 5.1 wt% at 100 bar and 77 K compared to 5.0 wt% for the UiO-66 powder. Depending on the method used to compute the volumetric uptake, the densified UiO-66 attained unprecedented volumetric capacity at 77 K and 100 bar of up to 74 g L−1 (13 g L−1 at 298 K) compared to 29 g L−1 for the powder (6 g L−1 at 298 K) using a conventional method that takes into account the packing density of the adsorbents, or 43 g L−1 (compared to 35 g L−1 for the powder at 77 K and 100 bar) based on a method that uses both the single crystal and skeletal densities of MOFs. However, regardless of the difference in the calculated values according to the two methods, the concept of UiO-66 compaction for improving volumetric capacity without compromising gravimetric uptake is clearly proven in this study and shows promise for the achievement of hydrogen storage targets for a single material as set by the United States Department of Energy (DOE).
1. Introduction
The use of metal–organic frameworks (MOFs) in cryogenic hydrogen (H2) storage applications has shown great promise due to the high surface areas and large micropore volumes exhibited by MOFs such as MOF-5 and MOF-177, among others.1 The highest measured total gravimetric uptake of 16.4 wt% H2 was reported for the copper-based paddlewheel MOF NU-100 at 77 K at a moderate pressure of 70 bar.2 Coupled to the high H2 adsorption capacities that can be displayed, the physisorption-based adsorption mechanism also allows for fast kinetics for the uptake and release (desorption) of H2 molecules with minimal energy inputs, making MOFs good candidates for use in on-board H2 storage systems in fuel cell vehicles.3 Additionally, more recently there have been attempts to make the synthesis of MOFs relatively simple and inexpensive, and best practices involved in their synthesis, activation and characterisation have been outlined.4 Synthesis procedures may include microwave-assisted methods,5,6 solvent-free routes,7 and the use of MOF precursors derived from waste materials, e.g. use of terephthalic acid (H2BDC) derived from waste polyethylene terephthalate (PET) to synthesise Zr-based MOF (UiO-66) and Cr-MOFs (MIL-101 and MIL-53).8–10 The combination of favourable surface areas and micropore volumes for physisorption-based H2 storage, and potential green synthesis methods renders MOFs as being of high interest and promising for industrial applications.The potential use of MOFs for material-based on-board hydrogen storage systems requires the selection of candidate MOFs that meet certain universally accepted targets. An example of such targets for 2020, as offered by the United States Department of Energy (DOE), require that the system has a gravimetric uptake of 4.5 wt% H2 and a volumetric capacity of 30 g L−1 H2.11 While some MOFs have been reported to attain total gravimetric H2 uptake exceeding 10 wt%, their respective volumetric H2 capacities seldom reach 30 g L−1. Indeed, many different types of MOFs have been assessed in efforts to reach these targets such as in the study by Goldsmith et al.12 who explored the relationship between total gravimetric uptake and total volumetric H2 capacity for a large number of MOFs. It was found that most MOFs have relatively low total volumetric densities and only a few were within the 2017 DOE target at the time (5.5 wt%; 40 g L−1) for a single storage material. The results indicated that a maximum volumetric H2 capacity of about 55 g L−1 was reachable, but that any further changes in materials properties to increase the total gravimetric H2 uptake resulted in a reduction in the total volumetric H2 capacity. This means that the surface area of the best performing MOFs should be limited to those that do not compromise the total volumetric H2 storage capacity of the MOF. Therefore, for H2 storage applications using MOFs, the focus needs to shift towards the direction where both the total volumetric H2 storage capacities and surface areas are simultaneously improved while maintaining the crystal structure of the MOF material.
As an alternative, the compaction of powdered samples into shaped bodies such as monoliths has been implemented to improve the total volumetric H2 capacity of MOFs. However, in most cases this improvement in volumetric capacity is always accompanied by a reduction in gravimetric uptake.13–16 Most of the previous studies on gas adsorption showed that the total volumetric H2 capacity of adsorbed gas is related to the density of the adsorbent and that by using shaping strategies such as compaction, the relatively low densities of MOFs can be increased in order to improve their total volumetric H2 storage capacity.13–15 Indeed, a study by Ardelean et al.16 reported the compaction of MIL-101 (Cr) powder to achieve envelope densities close to the crystal density of MIL-101 (Cr) and the resultant “monoliths” were shown to have an impressive total volumetric H2 capacity of 40 g L−1.
The challenge, however, is that the application of external pressure onto nanostructured porous materials has been shown to typically result in the reduction of surface area and may also induce the amorphisation of MOF crystals.17–19 Another response of porous materials to external applied pressure includes anisotropic changes in crystal topology, such as seen in the MIL family of MOFs (e.g. MIL-53 and MIL-101),20 which may result in widening of pores. These structural changes can have a direct impact to gas adsorption and it is generally observed in MOFs that upon compaction, the surface area reduces and results in a diminished total gravimetric H2 uptake.17,18,21–23 In order to counteract this inverse relationship between gravimetric H2 uptake and compaction (i.e. densification), the use of highly robust MOFs which show high resistance to applied pressure is desirable for MOF-based materials that retain their initial surface area and micropore volumes upon compaction at high pressure.
The textural properties of MOFs may also be modified by applying simple adjustments to the MOF synthesis conditions.24–28 One of the extensively investigated methods is the synthesis of MOFs with defects or missing organic linkers in their crystal lattice, which results in the presence of open metal sites within the MOF crystal structure.29–33 At the open transition metal (TM) centres there exists an opportunity for enhanced TM to gas interactions that may improve gas uptake as described in previous studies.34–36 The compaction of MOFs has been shown to lead to increased volumetric H2 capacities and in this study we aim to investigate the effects of compaction at significantly higher pressures (∼700 MPa or 100000 psi) than previously reported for UiO-66,17 and possibly improve the volumetric H2 capacity for UiO-66 towards the DOE target of 30 g L−1.
2. Experimental
2.1. Reagents and chemicals
Zirconium tetrachloride (ZrCl4, Sigma Aldrich, 99.5%), 1,4-benzenedicarboxylic acid (BDC, Sigma Aldrich, 98%), N,N-dimethylformamide (DMF, Sigma Aldrich, 99.8%), formic acid (HCOOH, Sigma Aldrich, 95%) and dried acetone (Sigma Aldrich, 99.8%) were purchased and used without further purification.2.2. Preparation of UiO-66
UiO-66 crystals were synthesised, with minor modifications, using a previously reported method.27 Typically, to a 500 mL round-bottom flask, a 1:1 molar ratio of ZrCl4:BDC was dissolved ultrasonically in 300 mL DMF for 1 h. To the mixture a 100 molar equivalent of formic acid was then added as a modulator and the flask capped with a thick-walled balloon. The reaction mixture was heated and maintained at 120 °C for 5 h and thereafter cooled to room temperature. The resulting white product was collected by centrifugation and washed in dried acetone and after re-collection, the solid product was dried under vacuum at room temperature.
2.3. Compaction of UiO-66
In order to select the optimum process parameters for compaction, a screening experiment was conducted whereby ∼400 mg UiO-66 powder was compacted in a 13 mm diameter die (area = 113 mm2) at 2, 4, 6, 8, and 9 metric tonnes (i.e. ∼150, 290, 440, 590, and 665 MPa applied pressure) in a Specac Manual Hydraulic Press for 15 minutes. The highest applied compaction pressure where the crystallinity of UiO-66 was still maintained giving the highest packing density for UiO-66 was chosen for H2 uptake measurements up to a hydrogen pressure of 100 bar.2.4. Characterisation
Powder X-ray diffraction (PXRD) patterns of powdered and compacted UiO-66 were collected within the 2θ range of 3–90° using a Rigaku Ultima IV X-ray diffractometer with CBO technology using Ni-filtered Cu-Kα radiation of 0.154 nm (40 kV, 30 mA) at a scanning speed of 2° s−1. The PXRD pattern of the compacted UiO-66 was done on a 1 mm × 20 mm pellet and mounted onto the PXRD sample holder by thinning the edges so that it fits into the well of the sample holder. The surface morphology was analysed using an Auriga cobra Focused-Ion Beam Scanning Electron Microscope (FIB-SEM) where each sample was mounted on a carbon tape and coated with carbon (where necessary) prior to each analysis. Nitrogen sorption and desorption isotherms were obtained on a Micromeritics 3 Flex sorptometer at 77 K and relative pressure (p/po) of up to 1. Pore size distribution (PSD) curves were obtained using the Non-Local Density Functional Theory (NLDFT) model. Ultra-high purity grade (99.999%) nitrogen gas was used and each sample was degassed on a Micromeritics Smart VacPrep under vacuum (down to 10−7 bar) with heating up to 80 °C for 32 h prior to each sorption isotherm measurement. The thermal stabilities of pristine and composite materials were measured with a thermogravimetric analyzer (TGA) (Mettler, Toledo, TGA/SDTA 851e) on degassed samples. For each run, ∼10 mg sample was heated to 1000 °C at a ramp rate of 10 °C min−1 under 100 mL min−1 airflow. Excess hydrogen uptake measurements were obtained from 0–100 bar at 77 K or room temperature using 6.0 grade H2 (99.9999%), additionally purified by a molecular sieve filter, on a Hiden Isochema XEMIS intelligent gravimetric analyser. The measured excess H2 uptake was obtained under non-equilibrium (dynamic) conditions as time intervals at each H2 pressure were manually inserted prior to the analysis. The measurements were also corrected for buoyancy effects, where the skeletal density of each MOF sample was determined by Helium pycnometry. The total H2 uptake was calculated based on the Langmuir surface coverage of adsorbed H2 at specified pressures as shown in eqn (1). | (1) |
The total volumetric H2 capacity was calculated from the gravimetric data using two approaches. The most commonly reported approach uses the packing density of the MOF material to calculate the total volumetric capacity as shown in eqn (2) and (3). The second approach comes from a recent study by Ahmed et al.37 who highlighted some recommendations made by Parilla et al.38 where the total volumetric H2 capacity was calculated from gravimetric data (excess H2 uptake in wt%) using both the single crystal and skeletal densities of MOFs according to eqn (4).
| vθExc = θExc × dMOF | (2) |
| vθT = θT × dMOF | (3) |
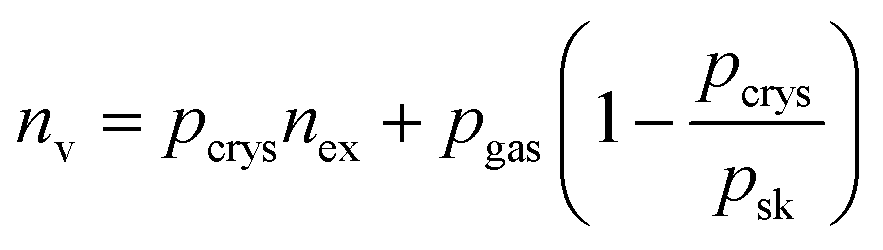 | (4) |
3. Results & discussion
The XRD pattern (Fig. 1a), and SEM image (Fig. S1, ESI†), respectively showed typical UiO-66 2θ peak positions (∼8, 9, and 25°) and octahedral crystal shapes as reported in previous studies.39–41 As can be seen in Fig. 1a there was very little difference in XRD patterns of compacted UiO-66 with increasing applied compaction pressure. The peak intensities, however, were found to slightly decrease and some evidence of peak broadening was also observed at high compaction pressure which may be caused by the onset of an amorphisation process (Fig. 1b). Nevertheless, the XRD patterns for UiO-66 compacted up to 665 MPa generally show the UiO-66 crystal structure was essentially retained upon compaction. The presence of the symmetry forbidden (1 1 0) reflection at the 2θ value of ∼6° was the first indication for the synthesis UiO-66 crystals with defects, i.e., crystals consisting of missing benzene dicarboxylate (BDC) organic linkers and hence the existence of open Zr centres.31 As further indication for defect UiO-66, TGA results (Fig. S2, ESI†) show that the complete structural collapse of the UiO-66 framework at ∼500 °C yielded a final residue of about 37 wt%, the composition of which is the non-porous solid ZrO2.42 Previous studies by Valenzano et al.,30 and Chavan et al.43 have demonstrated the use of TGA data to deduce the presence/absence of BDC linkers in UiO-66. Having found their as-prepared UiO-66 crystal structure to be made up of Zr6O4(OH)2(BDC)12 clusters, which undergo desolvation and dehydroxylation up to 300 °C to form Zr6O6(BDC)12 clusters, they showed that the final ZrO2 residue after framework collapse should yield no less than 45 wt% for perfect UiO-66 crystals with no defects. Final ZrO2 residues below 45 wt% would constitute one missing BDC linker per cluster, assuming an evenly distributed presence of defects. The TGA results obtained in this study also support a multi-step decomposition of UiO-66 between 90–300 °C before framework collapse at ∼500 °C, and these early mass loss events might involve the removal of DMF and H2O molecules by desolvation and dehydroxylation, respectively. Regarding thermal stability, we also found that the temperature at which thermal treatment (i.e., evacuation) was carried out prior to porosity determination had an influence on the BET surface area and porosity of UiO-66. The as-prepared UiO-66 samples heat treated under vacuum at 80 °C for 32 h showed higher BET surface area and pore volume than samples heat treated at 200 °C for 8 h under vacuum (Fig. S3, ESI†). This result may suggest the existence of UiO-66 polymorphic forms (hydroxylated and dehydroxylated forms) with slightly different gas adsorption properties in a manner similar to the findings reported by Chavan et al.43 and others.30,32 | ||
| Fig. 1 PXRD patterns of UiO-66 at increasing compaction pressure (0–665 MPa): (a) full patterns showing all peak positions for UiO-66, (b) section of the patterns from 2θ = 3–20° showing no changes to peak positions with increasing applied pressure. | ||
In Fig. 2a the N2 adsorption isotherms for both powder and compacted UiO-66 show type I adsorption and in Table 1 it can be seen that the compacted UiO-66 showed a less than 2% decrease in BET surface area (1707 m2 g−1) compared to UiO-66 powder (1737 m2 g−1), and ∼5% reduction in micropore surface area and micropore volume. Such a result is unusual for MOFs, as given the nature of their highly porous crystal structure, it can be expected that they show high compressibility, and negative response to applied pressure. Wu et al.,44 have reported that the minimum shear modulus for UiO-66 is ca. 13.7 GPa due to the strong Zr–O bonding in its crystal lattice, and is much higher than the value for most MOFs such as ZIF-8 (ca. 2.7) and MIL-53 (Ga) (ca. 0.16 GPa).45 Therefore, due to the little changes in crystal structure at ∼665 MPa, it can be expected that UiO-66 crystals would retain most of their textural properties at applied compaction pressures below the minimum shear modulus, as shown in this study. The total pore volume (Table 1), on the other hand, was found to differ more significantly with UiO-66 powder having 0.96 cm3 g−1 whereas the UiO-66 pellet was 0.81 cm3 g−1, a 16% reduction in total pore volume. This can be attributed to the significant reduction in macropores that are due to interparticle voids that are removed upon compaction as a result of the close packing of the particles (Fig. S4, ESI†).
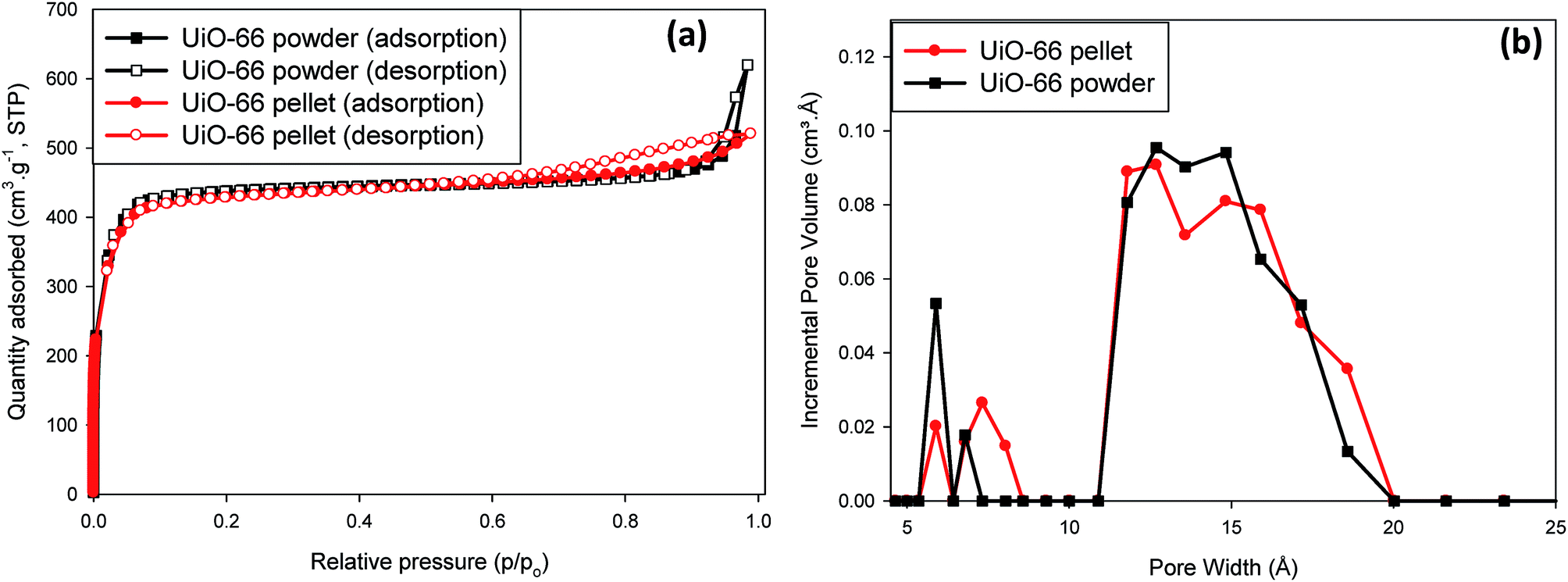 | ||
| Fig. 2 Textural properties for UiO-66 before and after compaction at 665 MPa: (a) N2 isotherms at 77 K; (b) pore size distribution in the micropore region (PSD < 20 Å). | ||
| Sample | Surface areaa (m2 g−1) | Pore volumeb (cm3 g−1) | Packing density (g cm−3) | Skeletal density (g cm−3) | Gravimetric H2 uptake (wt%) | Volumetric H2 capacity (g L−1) | Totalf | ||
|---|---|---|---|---|---|---|---|---|---|
| Excess | Total | Excessd | Totale | ||||||
| a Values in parenthesis are micropore surface area and percentage micropore surface area of the total surface area. b Values in parenthesis are micropore volume and percentage micropore of the total pore volume. c Tapped density of UiO-66 powder. d Excess volumetric capacity calculated from the packing density as per eqn (2). e Total volumetric capacity calculated from the packing density as per eqn (3). f Total volumetric H2 capacity calculated using the single crystal (1.24 g cm−3) and skeletal densities of UiO-66 as reported by ref. 37. | |||||||||
| UiO-66 powder | 1737 (1559, 90%) | 0.96 (0.60, 63%) | 0.57c | 1.65 | 2.1 (0.4) | 5.0 (1.1) | 12 (2) | 29 (6) | 35 (7) |
| UiO-66 pellet | 1707 (1484, 87%) | 0.81 (0.57, 70%) | 1.45 | 1.78 | 2.7 (0.3) | 5.1 (0.9) | 39 (4) | 74 (13) | 43 (6) |
The pore size distribution curves shown in Fig. 2b are consistent with reported literature with pores of size ∼6, 8 and 11 Å, which correspond to the free diameters in tetrahedral cages, triangular windows and octahedral cages of UiO-66, respectively.46,47 There was also evidence of a broadened pore size distribution towards higher values, with peaks at ∼14 Å up to ∼18 Å which was an additional sign of UiO-66 crystals with defects and possibly open Zr metal sites. The observation of retention of UiO-66 crystal structure after compaction up to 665 MPa as evident from the X-ray diffraction patterns in Fig. 1 was also translated in the textural properties of powdered and compacted UiO-66 forms (Fig. 2). A noticeable feature of the UiO-66 prepared in this study is that the surface area and pore volumes obtained are amongst the highest values reported for UiO-66 prepared using organic acid modulators.31,48 Shearer et al.31 reports an in-depth description on the role of monocarboxylic organic acids such as formic acid in the formation of defects in UiO-66, which results in increased BET surface areas compared to non-modulated (defect-free) UiO-66. The defects in crystals suggest the presence of open metal sites which can enhance the binding affinity of gas molecules at low gas pressures and may result in enhanced gas uptake than would be expected, especially within open metal centres contained in highly microporous environments such as found in MOFs.34,36
The trend in the total H2 uptake (wt%) data shown in Fig. 3a is consistent with that observed for the surface area for the powdered and compacted UiO-66. The powder and compacted UiO-66 have relatively similar hydrogen uptake, which means that there was no compromise of total H2 uptake after compaction of UiO-66 powder at 665 MPa pressure. There were, however, slight differences in their adsorption isotherms as it can be seen that the excess H2 uptake in UiO-66 powder reaches a maximum at relatively low pressure (20–30 bar) after which there is a decrease up to 100 bar. This is the typical behaviour in physisorption-based gas uptake due to rapid adsorption onto the adsorbent surface at low surface coverage (high number of available binding sites) followed by a higher desorption to adsorption rate upon saturation of the surface excess (i.e. excess surface coverage ≈ 1).49 In the excess H2 isotherm for compacted UiO-66, the pressure at which maximum uptake is reached was much higher (>50 bar), and also there was no significant decrease up to 100 bar. The uptake of the H2 is reversible (Fig. S5, ESI†) as the adsorption and desorption isotherms overlap. This is the expectation for physisorption based uptake and is also an indication of the robustness of our H2 sorption measurements.
 | ||
| Fig. 3 Gravimetric H2 uptake (wt%) for powder and compacted UiO-66 up to 100 bar measured at (a) 77 K and (b) 298 K. The plots are for excess (open symbols) and total (closed symbols) H2 uptake. | ||
Due to the compaction, it can be expected that access to binding sites/pores for H2 molecules would be more difficult due to the significant reduction in voids or macropores in-between UiO-66 particles resulting in a reduced grain boundary between the UiO-66 crystallites. As a result, the rate at which H2 molecules diffuse into the UiO-66 framework becomes reduced and higher pressures are required to reach maximum excess surface coverage and also the rate of desorption of H2 molecules within the pores becomes restricted by the close packing. The H2 adsorption at 298 K up to 100 bar in Fig. 3b were found to be higher for the UiO-66 powder compared to compacted UiO-66. As expected, physical adsorption of H2 in porous materials becomes lowered as the temperature increases, and higher pressures are required to observe improved adsorption.49 The difference observed for powdered and compacted UiO-66 can be resulting from kinetic effects as the diffusion of H2 in compacted UiO-66 would be slower compared to UiO-66 powder and because of the significantly reduced H2 adsorption at 298 K compared to 77 K, the excess H2 adsorption in compacted UiO-66 would only match that of powdered UiO-66 at pressures higher than 100 bar. The respective uptake values given in Table 1, comparing UiO-66 powder and UiO-66 pellet, show that at least 90% of the UiO-66 textural properties are retained upon compaction at 665 MPa applied pressure; the major difference is in their packing and skeletal densities, which influence the calculation of the total volumetric H2 capacity as given in eqn (3) and (4). As expected the packing density increased after compaction, however, it was interesting that the skeletal density also increased. The UiO-66 prepared in this study was a polycrystalline powder made up of different crystallite sizes (Fig. S1, ESI†). The compaction of UiO-66 powder at sufficiently high pressure would thus result in the reduction of the grain boundary between the crystallites, reducing its volume in the densified state. This may result in some micropores to be inaccessible under non-equilibrium gas uptake measurements such as those used for helium pycnometry and result in higher skeletal density results.
For the purpose of this study, the volumetric H2 capacity was calculated using two methods. Fig. 4a and b shows data obtained using eqn (4) taking into account the recommendations made by Parilla et al.38 for calculations based on gravimetric measurements of H2 uptake at high pressure. In Fig. 4a, the volumetric H2 capacity (at 77 K) for compacted UiO-66 exceeds that of powdered UiO-66 from 25–100 bar by up to ∼26%. Given that both the textural properties and the total gravimetric H2 uptake up to 100 bar were not compromised after compaction, the significant difference in volumetric H2 capacity can therefore be attributed to the improvement of the UiO-66 density after compaction. Indeed previous studies13,15,16 have largely reported on the improvement of volumetric H2 capacity of MOFs due to compaction but there have been seldom reports where the MOF gravimetric H2 uptake is also not compromised with compaction. Furthermore, the ratio of the volume of H2 adsorbed to the volume of H2 in the gaseous/bulk phase (Vad/Vbulk) can serve as an indication of the efficiency of H2 storage in the MOF compared to that of an empty container/cylinder. Under the specified isotherm conditions (T = 77 K, 0 < P < 100 bar), the H2 density (g L−1) increases linearly with pressure and Vad/Vbulk ≈ 1 as calculated using the ideal gas law, i.e., no adsorption would be expected in an empty container. In the presence of UiO-66, as shown in Fig. S6 (ESI†), the Vad/Vbulk ratio rapidly increases at pressures below 5 bar peaking at 80 cm3 cm−3 and 40 cm3 cm−3 for powdered and compacted UiO-66, respectively. This means that at pressures below 5 bar, UiO-66 stores at least 40 times more H2 by volume compared to the amount stored in an empty container/cylinder under the same conditions. As the pressure increased from ∼5 bar up to 100 bar, the Vad/Vbulk ratio decreases sharply initially and then steadily towards Vad/Vbulk ≈ 1. The rapid increase in adsorbed H2 at low pressure can be attributed to the readily available adsorption sites (such as pores and pore walls). As these spaces/sites become filled with mono- and/or multilayers of H2 molecules, the uptake of more H2 becomes restricted and may lead to an equilibrium state of adsorption being reached (i.e., saturation). Interestingly, from ∼30 to 100 bar, Vad/Vbulk for compacted UiO-66 is greater than that of the powdered UiO-66, showing more uptake of H2 at elevated pressures. This correlates with the excess adsorption isotherm obtained for compacted UiO-66 in relation to the UiO-66 powder, i.e., the densified sample takes up more excess H2. Clearly, the significant increase in the UiO-66 density after compaction at 665 MPa translates to a rise in the calculated total volumetric H2 capacity. Similar to the gravimetric H2 uptake, the volumetric capacities were also found to be significantly lower at 298 K compared to 77 K, and the UiO-66 powder had slightly higher uptake capacity than compacted UiO-66. This observation suggests that the adsorption onto compacted UiO-66 is significantly affected by kinetic effects under non-equilibrium H2 uptake conditions. Therefore, given that the surface area and pore volumes are effectively retained after compaction, it can be expected that at higher H2 adsorption pressures (greater than 100 bar), the room temperature volumetric hydrogen uptake capacity of compacted UiO-66 would exceed that of UiO-66 powder in a manner similar to that observed at 77 K.
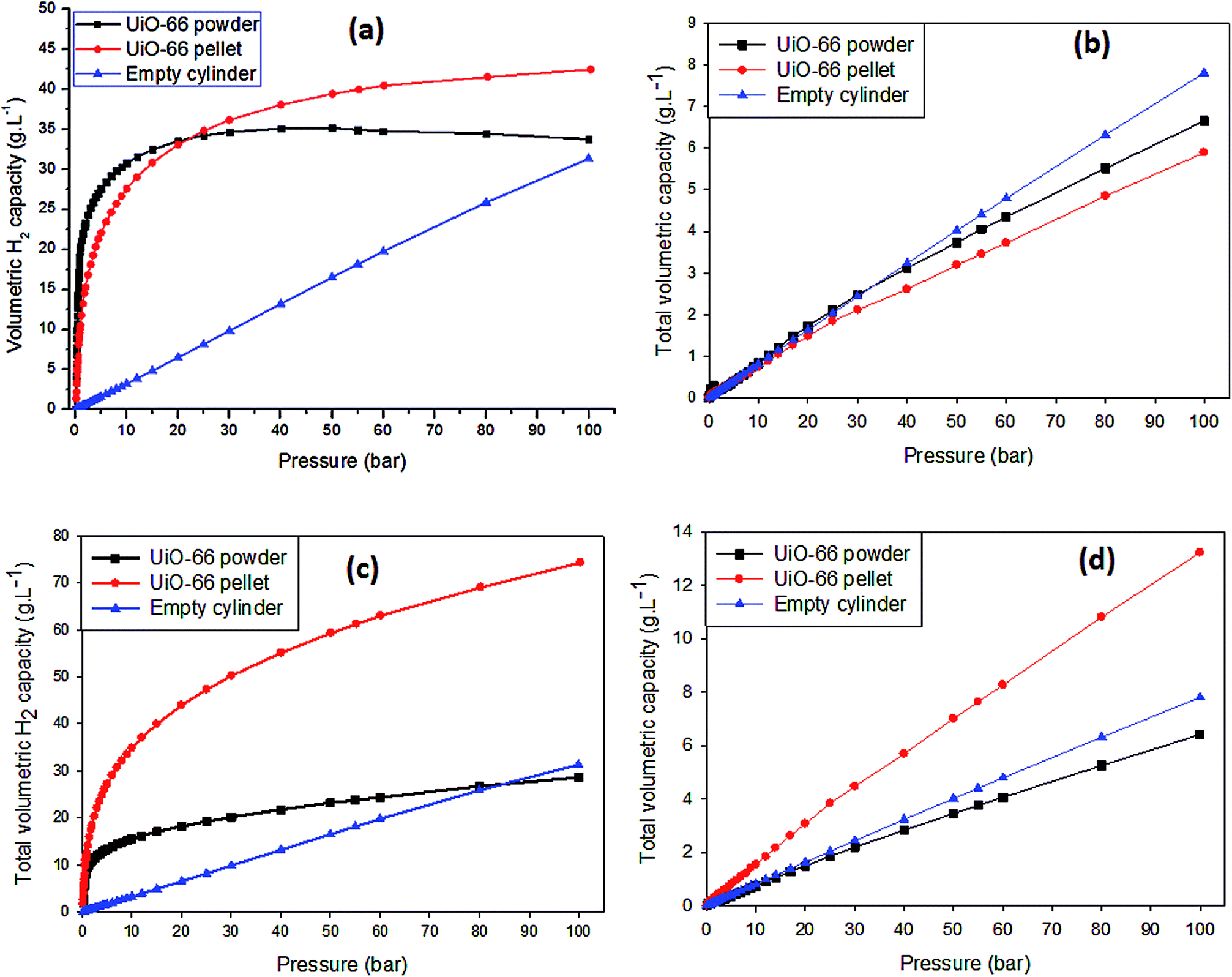 | ||
| Fig. 4 Volumetric H2 isotherms for powdered and compacted UiO-66 up to 100 bar: (a and b) volumetric H2 capacity/density (g L−1) at 77 K and 298 K obtained using eqn (4), and (c and d) volumetric H2 capacity/density at 77 K and 298 K obtained using eqn (3). | ||
We also report in Table 1 and Fig. 4c, d the total volumetric H2 capacity calculated using the packing density of powdered and compacted UiO-66, which is currently the most commonly used method of calculating the total volumetric capacity.13,16,50–53 The results show that there is a remarkable increase in total volumetric H2 capacity from 29 to 74 g L−1 (6 to 13 g L−1 at 298 K) due to the improved packing density of UiO-66 after compaction at 665 MPa as calculated using eqn (3). The results however, do show a significant improvement of the total volumetric capacity due to compaction regardless of the mathematical model for conversion of gravimetric data into volumetric values.
We also take note of the results obtained at 77 K and 25 bar (Table S2, ESI†) based on a recent study by Balderas-Xicohténcatl et. al.55 showing that the volumetric H2 capacity in MOFs follows a linear relationship with the MOF's volumetric surface area (m2 mL−1), similar to Chahine's rule. In our results, the compaction of UiO-66 powder increased the packing density from 0.57 to 1.45 g mL−1 and thus increasing the volumetric surface area from 990 to 2475 m2 mL−1. We see that multiplying the volumetric surface area by the slope 1.9 × 10−2 m2 mL−1 (derived from calculations made by Balderas-Xicohténcatl et. al.55) we obtain a calculated theoretical total volumetric capacity of 18 and 47 mg mL−1 (g L−1) for powder and compacted UiO-66, respectively. It is noteworthy that such a calculation yields virtually identical total volumetric capacities at 77 K and 25 bar, as calculated in our work described above using the packing density in eqn (3), for both powder and compacted UiO-66. The calculations and derivations made by Balderas-Xicohténcatl and co-workers were based on many different types of MOFs and also took into consideration the effect of compacting MOFs to increase packing densities. The strong agreement between our results to their estimations serves to validate our data and calculations and moreover, more generally, a strong indicator of the significant influence of packing density on volumetric H2 capacity in MOF powders and other related porous materials.
Conclusion
The aim of the study was an attempt at improving the total volumetric H2 capacity of UiO-66 without significantly reducing the gravimetric H2 uptake. Compaction of UiO-66 powder at 2, 4, 6, 8, and 10 metric tonnes (150, 290, 440, 590, and 665 MPa) did not lead to structural collapse of the UiO-66 crystal lattice. The compaction of MOFs at pressures as high as 665 MPa (∼97000 psi), without framework collapse, is unprecedented according to our knowledge as most previous studies have reported lower applied compaction pressures.13–23 The prepared UiO-66 had a surface area and total pore volume of ∼1700 m2 g−1 and 1.0 cm3 g−1, respectively. Furthermore, the surface area and micropore volume were retained to within at least 95% after compaction at 665 MPa. This non-changing porosity was translated in the total H2 uptake (at 77 K) with powdered and compacted UiO-66 having 5.0 and 5.1 wt%, respectively, at 100 bar. Given that the measurement techniques for surface area and H2 uptake were independent of each other, the results show an unambiguous outcome that clearly demonstrates the retention of textural and adsorption properties for UiO-66 upon compaction at ∼700 MPa. The compaction also predictably improved the UiO-66 density, with the skeletal and packing densities increasing from 1.65 to 1.78 g cm−3 and 0.57 to 1.45 g cm−3, respectively. The total volumetric H2 capacity was higher for the compacted UiO-66 than powdered UiO-66, regardless of the model/method of calculation utilized to convert gravimetric data into volumetric values. Using the model developed by Ahmed et al.,37 we obtained a volumetric H2 capacity of 43 g L−1 (100 bar, 77 K) and 6 g L−1 (100 bar, 298 K) for compacted UiO-66 compared to 35 g L−1 (100 bar, 77 K) and 6 g L−1 (100 bar, 298 K) for powdered UiO-66. Using the packing densities, which is the most commonly reported method for calculations, the volumetric H2 capacity at 100 bar was calculated to be 74 g L−1 (13 g L−1 at 298 K) and 29 g L−1 (6 g L−1 at 298 K) for compacted and powdered UiO-66, respectively. Even though the calculated values of total volumetric H2 capacity differ depending on the model used, the results however, clearly show an improvement in volumetric capacity upon compacting UiO-66. The results obtained in this study show that the fabrication of MOFs using techniques such as compaction can serve as a basis for developing MOF-based materials that can satisfy both gravimetric and volumetric US DOE targets for a single material.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge financial support from the Royal Society – DFID Africa Capacity Building Initiative Programme (Grant No. AQ150029), and the South African Department of Science and Technology (DST) towards HySA Infrastructure (Project No. EIMH01X).References
- M. P. Suh, H. J. Park, T. K. Prasad and D. Lim, Chem. Rev., 2017, 112, 782–835 CrossRef PubMed.
- O. K. Farha, A. O. Yazaydin, I. Eryazici, C. D. Malliakas, B. G. Hauer, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944 CrossRef CAS PubMed.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39(2), 656–675 RSC.
- A. J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 19, 26–39 CrossRef.
- J. Klinowski, F. A. Almeida Paz, P. Silva and J. Rocha, Dalton Trans., 2011, 40(2), 321–330 RSC.
- J. Ren, S. Segakweng, H. W. Langmi, N. M. Musyoka, B. C. North, M. Mathe and D. Bessarabov, Int. J. Mater. Res., 2014, 105(5), 516–519 CrossRef CAS.
- L. Paseta, G. Potier, S. Sorribas and J. Coronas, ACS Sustainable Chem. Eng., 2016, 4(7), 3780–3785 CrossRef CAS.
- W. P. R. Deleu, I. Stassen, D. Jonckheere, R. Ameloot and D. E. De Vos, J. Mater. Chem. A, 2016, 4(24), 9519–9525 RSC.
- X. Dyosiba, J. Ren, N. M. Musyoka, H. W. Langmi, M. Mathe and M. S. Onyango, Sustainable Mater. Technol., 2016, 10, 10–13 CrossRef CAS.
- J. Ren, X. Dyosiba, N. M. Musyoka, H. W. Langmi, B. C. North, M. Mathe and M. S. Onyango, Int. J. Hydrogen Energy, 2016, 41, 1841–1846 Search PubMed.
- Technical System Targets: Onboard Hydrogen Storage for Light-Duty Fuel Cell Vehicles, cited on 29 June 2018 from: https://www.energy.gov/sites/prod/files/2017/05/f34/fcto_myrdd_table_onboard_h2_storage_systems_doe_targets_ldv_1.pdf.
- J. Goldsmith, A. G. Wong-Foy, M. J. Cafarella and D. J. Siegel, Chem. Mater., 2013, 25(16), 3373–3382 CrossRef CAS.
- J. J. Purewal, D. Liu, J. Yang, A. Sudik, D. J. Siegel, S. Maurer and U. Müller, Int. J. Hydrogen Energy, 2012, 37(3), 2723–2727 CrossRef CAS.
- R. Zacharia, D. Cossement, L. Lafi and R. Chahine, J. Mater. Chem., 2010, 20(11), 2145–2151 RSC.
- G. Blanita, I. Coldea, I. Misan and D. Lupu, Int. J. Hydrogen Energy, 2014, 39(30), 17040–17046 CrossRef CAS.
- O. Ardelean, G. Blanita, G. Borodi, M. D. Lazar, I. Misan, I. Coldea and D. Lupu, Int. J. Hydrogen Energy, 2013, 38(17), 7046–7055 CrossRef CAS.
- G. W. Peterson, J. B. Decoste, T. G. Glover, Y. Huang, H. Jasuja and K. S. Walton, Microporous Mesoporous Mater., 2013, 179, 48–53 CrossRef CAS.
- S. A. Moggach, T. D. Bennett and A. K. Cheetham, Angew. Chem., Int. Ed., 2009, 48(38), 7087–7089 CrossRef CAS PubMed.
- D. Bazer-Bachi, L. Assié, V. Lecocq, B. Harbuzaru and V. Falk, Powder Technol., 2014, 255, 52–59 CrossRef CAS.
- F. X. Coudert, Chem. Mater., 2015, 27(6), 1905–1916 CrossRef CAS.
- M. C. Raj, S. Senthilkumar, R. S. Somani and H. C. Bajaj, Int. J. Environ. Stud., 2016, 73(3), 357–368 CrossRef CAS.
- K. Chapman, G. Halder and P. Chupas, J. Am. Chem. Soc., 2009, 131, 17546–17547 CrossRef CAS PubMed.
- N. Chanut, A. D. Wiessum, U.-H. Lee, Y. K. Hwang, F. Ragon, H. Chvreau, S. Bourrelly, B. Kuchta, J.-S. Chang, C. Serre and P. L. Llewellyn, Eur. J. Inorg. Chem., 2016, 2016(27), 4416–4423 CrossRef CAS.
- J. Ren, N. M. Musyoka, H. W. Langmi, T. Segakweng, B. C. North, M. Mathe and K. Xiangdong, Int. J. Hydrogen Energy, 2014, 39(23), 12018–12023 CrossRef CAS.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49(82), 9449 RSC.
- Q. Yang, A. D. Wiersum, P. L. Llewellyn, V. Guillerm, C. Serre and G. Maurin, Chem. Commun., 2011, 47(34), 9603 RSC.
- J. Ren, H. W. Langmi, B. C. North, M. Mathe and D. Bessarabov, Int. J. Hydrogen Energy, 2014, 39(2), 890–895 CrossRef CAS.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17(24), 6643–6651 CrossRef CAS PubMed.
- S. Øien, D. Wragg, H. Reinsch, S. Svelle, S. Bordiga, C. Lamberti and K. P. Lillerud, Cryst. Growth Des., 2014, 14(11), 5370–5372 CrossRef.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23(7), 1700–1718 CrossRef CAS.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28(11), 3749–3761 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26(14), 4068–4071 CrossRef CAS.
- T. K. A. Hoang and D. M. Antonelli, Adv. Mater., 2009, 21(18), 1787–1800 CrossRef CAS.
- C. Chung, J. Ihm and H. Lee, J. Korean Phys. Soc., 2015, 66(11), 1649–1655 CrossRef CAS.
- M. R. Andalibi, A. Qajar and H. C. Foley, J. Phys. Chem. C, 2015, 119(37), 21314–21322 CrossRef CAS.
- A. Ahmed, Y. Liu, J. Purewal, L. D. Tran, A. G. Wong-Foy, M. Veenstra, A. J. Matzger and D. J. Siegel, Energy Environ. Sci., 2017, 10(11), 2459–2471 RSC.
- P. A. Parilla, K. Gross, K. Hurst and T. Gennett, Appl. Phys. A: Mater. Sci. Process., 2016, 122(3), 1–18 CrossRef CAS.
- Y. Zhao, Q. Zhang, Y. Li, R. Zhang and G. Lu, ACS Appl. Mater. Interfaces, 2017, 9, 15079–15085 CrossRef CAS PubMed.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22(10), 6632–6640 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130(42), 13850–13851 CrossRef PubMed.
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46(41), 7700–7702 RSC.
- S. Chavan, J. G. Vitillo, O. Gianolio, O. Zavorotynska, B. Civalleri, S. Jakobsen, M. H. Nilsen, L. Valenzano, C. Lamberti, K. P. Lillerud and S. Bordiga, Phys. Chem. Chem. Phys., 2012, 14(5), 1614–1626 RSC.
- H. Wu, T. Yildirim and W. Zhou, J. Phys. Chem. Lett., 2013, 4(6), 925–930 CrossRef CAS PubMed.
- A. U. Ortiz, A. Boutin, A. H. Fuchs and F. Coudert, Phys. Lett., 2012, 109, 195502 CrossRef PubMed.
- G. W. Peterson, S.-Y. Moon, G. W. Wagner, M. G. Hall, J. B. DeCoste, J. T. Hupp and O. K. Farha, Inorg. Chem., 2015, 54, 9684–9686 CrossRef CAS PubMed.
- C. H. Lau, R. Babarao and M. R. Hill, Chem. Commun., 2013, 49, 3634–3636 RSC.
- K. Wang, C. Li, Y. Liang, T. Han, H. Huang, Q. Yang, D. Liu and C. Zhong, Chem. Eng. J., 2016, 289, 486–493 CrossRef CAS.
- K. M. Thomas, Catal. Today, 2007, 120(3–4), 389–398 CrossRef CAS.
- H. Furukawa, M. A. Miller and O. M. Yaghi, J. Mater. Chem., 2007, 17(30), 3197–3204 RSC.
- A. Dailly and E. Poirier, Energy Environ. Sci., 2011, 4(9), 3527–3534 RSC.
- L. Zhou, Renewable Sustainable Energy Rev., 2005, 9(4), 395–408 CrossRef CAS.
- G. Ye, H. Xin, Z. Bao-lin, M. Bao-guo and Z. Hong-bo, Powder Technol., 2008, 187(1), 88–93 CrossRef CAS.
- Thermophysical properties of fluid systems, Cited on 15 February 2018 from: http://www.nist.gov.
- R. Balderas-Xicohténcatl, M. Schlichtenmayer and M. Hirscher, Energy Technol., 2018, 6, 578–582 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Six additional figures (consisting of SEM images, TGA analysis, comparative nitrogen isotherms and pore size distribution curves, and adsorbed H2 volume fractions), and two tables (consisting of comparative textural properties, packing density, and H2 uptake). See DOI: 10.1039/c8ta09227c |
| This journal is © The Royal Society of Chemistry 2018 |