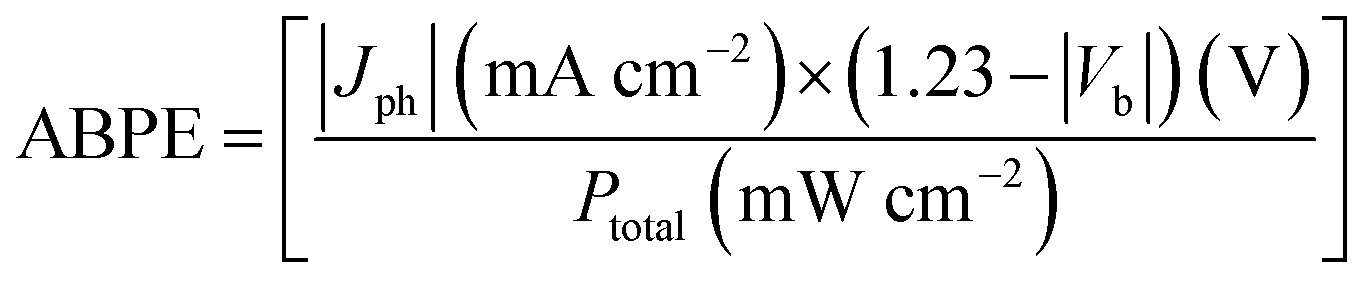DOI:
10.1039/C8TA08631A
(Paper)
J. Mater. Chem. A, 2018,
6, 24358-24366
Atomically manipulated proton transfer energizes water oxidation on silicon carbide photoanodes†
Received
5th September 2018
, Accepted 13th November 2018
First published on 13th November 2018
Abstract
Surmounting the sluggish water oxidation kinetics beyond the hole-dominated thermodynamic effect is a topic of great scientific interest to establish fully renewable hydrogen technology from solar-powered water splitting. Herein, we demonstrate that the bottleneck of photoelectrochemical water oxidation can be overcome via atomic manipulation of proton transfer on the polar surfaces of silicon carbide (SiC) photoanodes. On the typical carbon-face SiC, where proton-coupled electron transfer governed the interfacial hole transfer for water oxidation, substantial energy loss was inevitable due to the highly activated proton-transfer steps. Via preferentially exposing the silicon-face, we enabled surface-catalyzed barrierless O–H breaking with a facile proton exchange and migration character. This mechanistically shifted the rate limiting step of water oxidation from sluggish proton-coupled electron transfer to a more energy-favorable electron transfer. The proof-of-concept study introduced here may open up new possibilities to design sophisticated photoelectrodes for an unbiased solar water splitting cell via surface engineering.
1. Introduction
Solar energy can provide sufficient power to meet the world's energy demand if it can be efficiently harvested. This is the very reason why photoelectrochemical (PEC) water splitting and CO2 reduction attract worldwide research interest as a promising sustainable technology for solar fuel production.1–4 The PEC conversion of solar energy into chemical fuels is based on two half-reactions, namely, water oxidation and reduction reactions. The slow-rate oxygen evolution reaction (OER) at photoanodes in many cases is regarded as the bottleneck for PEC reactions, especially when photoanodes are coupled to many metals which can achieve a hydrogen evolution reaction at relatively low overpotentials. This is because the OER involves a four-electron process via several steps with high energy barriers (2H2O → O2 + 4H+ + 4e−).5,6 Despite the large number of methodologies which have been aiming to reduce the OER overpotential, including doping, surface modification, and nanostructure tuning, it still remains a challenge to design a highly active photoanode because the detailed mechanisms of the OER kinetics are still not fully understood.
Intrinsically, it is generally agreed that the OER in a typical PEC reaction is composed of the following four sequential proton-coupled electron transfer (PCET) steps:7–10
| | | *O + H2O(l) → *OOH + H+ + e− | (3) |
where * denotes the surface adsorbed state, e
− is the interfacial electron, H
2O(l) is the liquid water, and H
+ is the dissociated proton. Among them, the first PCET (*H
2O → *OH + H
+ + e
−), being characterized by both O–H bond breaking associated proton-transfer (PT) and interfacial electron-transfer (ET), is envisioned as the rate-determining step.
10 The first PCET of the OER has long been believed to be governed by ET, whose reaction thermodynamics is highly dependent on the redox level of the hole in the valence band (VB) of the photoanode. However, it cannot explain the ineffectiveness of the OER on many pristine semiconductors, including TiO
2, Fe
2O
3 and ZnO, whose VB positions perfectly match with that of water oxidation.
11,12 The pioneering work done by Selloni and Liu groups, on the other hand, theoretically demonstrated that beyond the hole-dominated thermodynamic effect, strong kinetic effects concerned with PT were also essential for the OER.
10,13–15 For instance, the first PCET was demonstrated to be sequential with PT (*H
2O → *OH
− + H
+) preceding ET (*OH
− → *OH + e
−); besides, an inherent high overpotential had arisen from highly activated O–H oxidative cleavage and proton migration within the PT step, rather than the barrierless ET. As a proof of concept, this theory is consistent with the abundant theoretical and experimental findings that thermodynamic hole trapping by water was remarkably enhanced at high pH, when the proton concentration of the electrolyte was low and only deprotonated surface states possessed appreciable energy to trigger the OER, all of which offer a possibility to accelerate the sluggish OER
via modifying the delicate PT behavior.
16–19
Silicon carbide (SiC) has emerged as a promising semiconductor for photocatalytic applications due to its superior chemical stability, high charge mobility, and suitable energy band positions that straddle the water redox potential.20,21 Most noticeably, SiC is receiving considerable attention as a novel biocompatible material due to its tunable surface water chemistry, particularly for the representative polar (carbon or silicon terminated) surfaces, which are commercially available. Depending on the terminated atoms (C or Si), the exposed surfaces display different atomic and electronic structures, which remarkably influence water adsorption, hydroxylation and wet oxidation behaviors. For example, the Si-face of SiC was theoretically proved to be capable of inducing water dissociation towards the formation of surface Si–OH and Si–H fragments, while the C-face displayed “inertness” to afford only molecular water adsorption, as demonstrated by Cicero and Amy et al.22–24 In the light of this, the polar surfaces of SiC have emerged as an ideal platform to manipulate PT behavior for desirable water oxidation and understand more of the details of water oxidation mechanisms. In this study, we theoretically and experimentally demonstrate that the bottleneck of the OER can be overcome via atomic manipulation of PT over the SiC polar surfaces. By preferentially exposing the Si-face, we enabled surface-catalyzed barrierless O–H breaking and proton exchange and migration within the PT step in the first proton removal reaction, which mechanistically shifted the sluggish rate-limiting step on the C-face to a more energy-favorable one, thus making the OER conductive at a very low overpotential even under neutral pH conditions. Mechanistic insights into the challenge of the PT scheme are provided on the basis of detailed theoretical and experimental results.
2. Results and discussion
2.1 Theoretical study of water adsorption on SiC
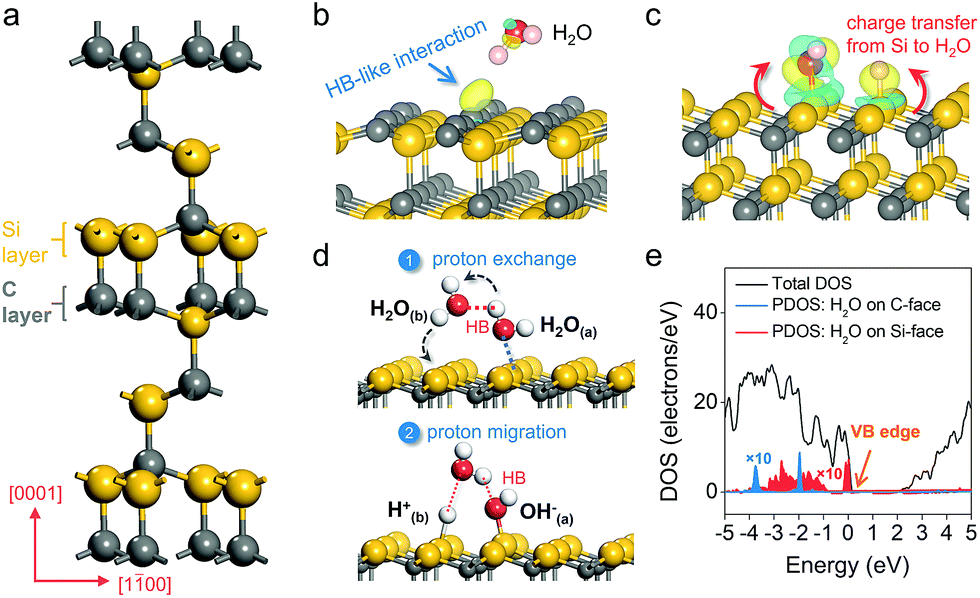
Density functional theory calculation was first used to model water adsorption and reaction on a prototypical SiC polar surface, the hexagonal 6H–SiC(0001) surface (Fig. 1a). On the C-terminated (000![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) surface, H2O was found to remain floating above the surface at low coverage (1/4 ML); even at higher coverage (when two H2O molecules were included into a supercell), water molecules preferred to optimize the internal hydrogen bond (HB) network without interacting with the C-surface (Fig. 1b). The charge density difference revealed an extremely weak HB-like interaction, as reflected by the appearance of charge localization in the region between a surface carbon atom and the –OH group of water. An entirely different picture of water adsorption was found when the Si-terminated (0001) surface was exposed. Irrespective of its initial orientation, H2O would diffuse to a Si–Si dimer, where spontaneous dissociation took place (H2O → OH− + H+) with the –OH fragment bonding to one Si atom, and the –H fragment being transferred to another Si atom (Fig. 1c). Such water dissociation behavior was due to a thermodynamic need to stabilize the dangling bonds and minimize surface energy,23 as evidenced by the significant charge depletion on the Si–Si dimer and charge accumulation on dissociated water fragments with a large adsorption energy of −3.36 eV (Fig. 1c).
) surface, H2O was found to remain floating above the surface at low coverage (1/4 ML); even at higher coverage (when two H2O molecules were included into a supercell), water molecules preferred to optimize the internal hydrogen bond (HB) network without interacting with the C-surface (Fig. 1b). The charge density difference revealed an extremely weak HB-like interaction, as reflected by the appearance of charge localization in the region between a surface carbon atom and the –OH group of water. An entirely different picture of water adsorption was found when the Si-terminated (0001) surface was exposed. Irrespective of its initial orientation, H2O would diffuse to a Si–Si dimer, where spontaneous dissociation took place (H2O → OH− + H+) with the –OH fragment bonding to one Si atom, and the –H fragment being transferred to another Si atom (Fig. 1c). Such water dissociation behavior was due to a thermodynamic need to stabilize the dangling bonds and minimize surface energy,23 as evidenced by the significant charge depletion on the Si–Si dimer and charge accumulation on dissociated water fragments with a large adsorption energy of −3.36 eV (Fig. 1c).
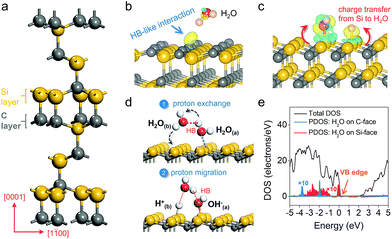 |
| | Fig. 1 Theoretical investigation of water adsorption and PT behavior on the C- and Si-polar surfaces of hexagonal 6H–SiC. (a) Unit cell of 6H–SiC. Adsorption of H2O on the (b) C-surface and (c) Si-face of 6H–SiC. The yellow and blue isosurfaces represent charge accumulation and depletion in the space along with water adsorption, respectively. The isovalue is 0.005 au. (d) Proton exchange and migration on the Si-face. Red dashed lines show the ordinary hydrogen bond (HB) or hydrogen-like bond. (e) Partial density of states (PDOS) of water on the Si-face and C-face of 6H–SiC with different adsorption structures. For the sake of presentation, the local contributions have been scaled by a factor of 10. | |
To explore the explicit first shell solvation effect, water adsorption at higher coverage was then modeled. When two H2O molecules were included into a supercell, we observed an interesting synchronous two proton exchange and migration phenomenon. Specifically, besides establishing a Lewis acid-base interaction with the Si surface, H2O(a) formed a reliable HB with a nearby H2O(b). Along with H2O(a) dissociation, the –Ha fragment, that was initially hydrogen-bonded, was transferred to H2O(b), while H2O(b) simultaneously released a –Hb fragment to a neighboring Si atom (Fig. 1d). Besides intermolecular proton exchange, proton exchange with a surface hydroxyl group on the Si-face along with water dissociation was also thermodynamically favorable (Fig. S1a†). This interesting proton exchange and migration behavior remarkably promoted the proton transfer ability (from one Si–Si dimer to another Si–Si dimer) on the Si-terminated surface (Fig. 1d). When three H2O molecules were included into a supercell, two of them preferred to dissociate, while the third one formed a HB network with the two dissociated H2O molecules (Fig. S1b†). The HB network contained two strong HBs and one weak HB, representing a typical intimate interaction between a wetting surface with liquid water (Fig. S1c†). Moreover, the formation of the HB3 lengthened the Si–H bond from 1.50 Å to 1.74 Å, simultaneously reducing the barrier of proton transfer to liquid water from 0.85 eV to 0.32 eV. It has to be pointed out that when H2O was pushed to the C-face at a distance below 1.0 Å, it also dissociated on a C–C dimer. However, water dissociation on the C-face has to overcome a kinetic barrier of 1.41 eV, suggesting that such a reaction would not spontaneously occur even at higher temperature. To estimate the thermodynamics of water oxidation, we calculated the partial density of states (PDOS) of adsorbed water on different exposed surfaces of 6H–SiC. For H2O weakly bound to the C-face via a HB, its PDOS was deeply embedded in the VB, approximately 2.02 eV lower compared to the VB edge of 6H–SiC, suggesting that hole transfer to the surface adsorbed H2O was thermodynamically hindered (Fig. 1e). However, dissociated water on the Si-face was apparently more active than molecular water on the C-face towards oxidation, as evidenced by its PDOS that almost overlapped the VB edge (Fig. 1e).25–27 This is consistent with the fact that oxidation of OH− to O2 is thermodynamically more favorable than the oxidation of H2O to O2 (EO2/OH− = 0.40 V and EO2/H2O = 1.23 V versus the reversible hydrogen electrode).16
2.2 Experimental study of water adsorption on SiC
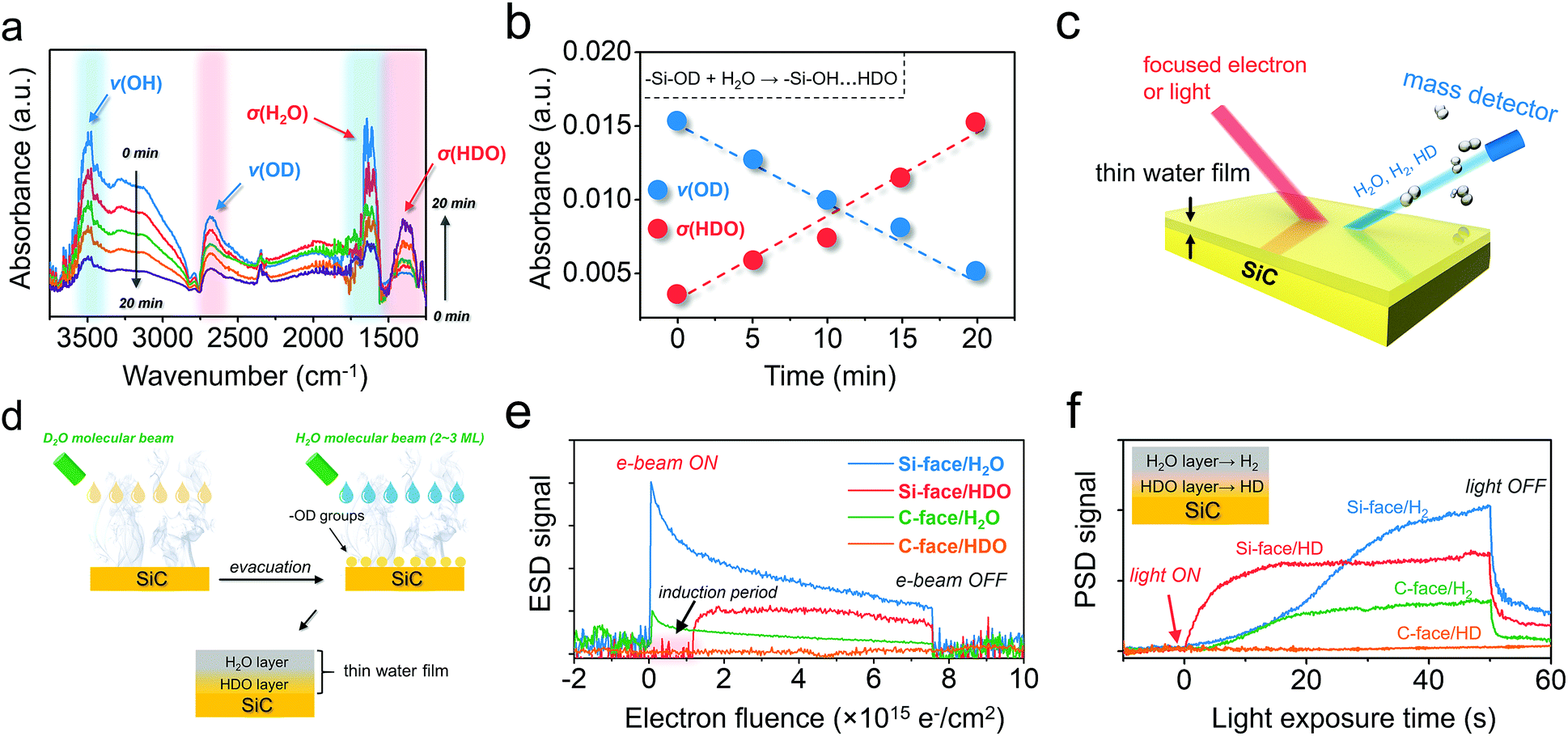
The above theoretical scenarios revealed that the Si-face on 6H–SiC not only enabled barrier-less O–H breaking on water adsorption, but also facilitated proton exchange and migration within the PT step, representing a key prerequisite for kinetically improved water oxidation. Meanwhile, from a thermodynamic point of view, water in the dissociated form (OH− + H+) should be more active with holes than its molecular form, suggesting the potential superiority of the Si-face for water oxidation. To experimentally verify these calculation results, n-type C-terminated and Si-terminated 6H–SiC films with device quality were grown on the C-face 6H–SiC(000) and Si-face 6H–SiC(0001) substrates by chemical vapor deposition (CVD), respectively.28 The surface morphology of the as-grown films was examined by atomic force microscopy (Fig. S2†). Before evaluating its PEC performance, specific water adsorption structures on both C- and Si-face 6H–SiC were investigated via Fourier-transform infrared (FTIR) spectroscopy in a designed reaction cell. The FTIR of pristine C- and Si-face 6H–SiC exhibited two weak absorption bands spanning 3600–3800 cm−1 ascribed to residual hydroxyl groups stretching with no contaminants like F− and NH3. To avoid the possible interference of these hydroxyl groups, heavy water (D2O)-saturated Ar gas was introduced to simulate water adsorption onto 6H–SiC surfaces. After sufficient D2O adsorption, the reaction cell was thoroughly evacuated (to ∼10−6 mbar) at 120 °C (the boiling point of D2O is 101.4 °C). No remarkable difference was observed on the C-face before and after D2O adsorption, suggesting that D2O was weakly interacting with the C-face and thermal heating allowed full desorption of D2O. In contrast, a strong ν(OD) stretching peak appeared at 2685 cm−1 (Fig. S3a†). The reason why the newly appeared absorption band at 2685 cm−1 was indexed to –OD groups arising from D2O dissociation (D2O → OD− + D+) could be explained by two facts. First, D2O was unlikely to exist on the Si face under continuous evacuation at 120 °C, as the boiling point of D2O is 101.4 °C. Second, with further increasing the heating temperature in the reaction cell to 250 °C, the strong absorption band at 2685 cm−1 experienced a slight decrease but was still clearly observed, suggesting that it should be attributed to strongly adsorbed –OD groups on SiC rather than weakly bound D2O (Fig. S3b†).26
To explore the possible proton exchange and migration scheme occurring on the Si-face, we further exposed the Si-face, which was pre-hydroxylated with –OD, to saturated H2O vapor. Following a transient water deposition at 298 K, the saturated H2O vapor in the reaction cell was then gradually re-evacuated with the surface hydroxyl stretching being in situ monitored. Along with evacuation, the broad absorption band ranging from 2700 to 3700 cm−1 and the band at 1628 cm−1, which were respectively assigned to ν(OH) and σ(H2O), gradually decreased (Fig. 2a).5 However, IR absorption of ν(OD), which was supposed to remain chemically and thermally stable, experienced a synchronous decrease, suggesting that part of the –OD on the Si-face was consumed. Unexpectedly, concomitant with the ν(OD) decrease, a new absorption band at 1400 cm−1 gradually appeared, which was indexed to the bending absorption of σ(HDO) (Fig. 2a).5 Since the IR absorption of the σ(HDO) region could maintain high stability under even higher vacuum, the as-formed HDO was believed to reside in the wetting layer, which interacted strongly with the hydroxyl groups on the Si-face via HBs (–Si–OH⋯HDO). Moreover, the almost linear decrease of ν(OD) and increase of σ(HDO) along with evacuation time suggested that the consumption of –OD on the Si-face was directly associated with HDO generation (Fig. 2b). Conceivably, facile proton exchange and migration between –OD on the Si-face and outer layer H2O was supposed to dominate this process (–Si–OD + H2O → –Si–OH⋯HDO) (Fig. S4a†).
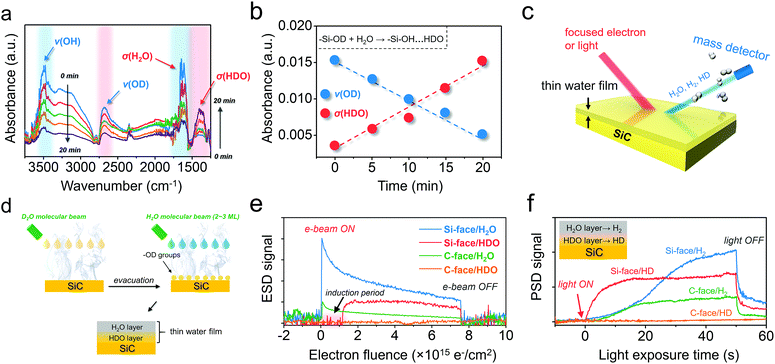 |
| | Fig. 2 Water adsorption behavior of the C- and Si-face of 6H–SiC. (a) Dynamic change of the FTIR spectra of the H2O-saturated Si-face with –OD hydroxylation under continuous evacuation. (b) Absorbance change of the band of ν(OD) and σ(HDO) along with evacuation time increase shown in (a). (c) Schematic illustration of the electron-stimulated or photon-stimulated desorption (ESD or PSD) technique for the investigation of water adsorption on 6H–SiC. (d) Preparation of a thin water film with a possible proton exchange layer for ESD and PSD analyses. (e) ESD signal of H2O and HDO versus electron fluence from the thin water film on the C- and Si-face 6H–SiC. (f) PSD signal of H2 and HD production from the thin water film on the C- and Si-face 6H–SiC. | |
In order to more precisely specify the components in the above water film on the Si-face, electron-stimulated and photon-stimulated desorption (ESD and PSD) techniques were carried out (Fig. 2c).29–32 In order to prepare a thin water film with a possible proton exchange layer on 6H–SiC, D2O was first deposited on SiC to ensure –OD hydroxylation, followed by evacuation (Fig. 2d). Then, 2–3 ML H2O was deposited on the hydroxylated 6H–SiC surfaces to enable the formation of a hybrid thin water film (Fig. 2d). On the Si-face of 6H–SiC, we were able to observe an abrupt H2O signal increase when the electron beam was turned on, followed by a rapid decay, indicating that stimulated H2O desorption directly happened on the water/vacuum interface (Fig. 2e). Besides, another signal corresponding to the proton exchange product of HDO was vividly witnessed on the Si-face, as depicted by a promotive increase but remarkably slower decrease (Fig. 2e). It is to be noted that an induction period appeared during stimulated HDO desorption. This is because electron-stimulated HDO desorption would occur only when the outer-layer H2O was largely desorbed (Fig. S4b†), confirming that the proton exchange and migration region was located at the water/6H–SiC interface. As for the C-face, it displayed an appreciably lower H2O desorption and no observable signal of HDO, in agreement with its poor affinity to water as will be shown below. We also considered the effect of light (λ < 400 nm) illumination on the desorption manners of other related molecules, including H2 and HD. After the light was turned on, we successfully observed the desorption of H2 from the Si-face or C-face, arising from water splitting, which was pronounced, but sluggish, as it was saturated only after 40 s (Fig. 2f). Interestingly, another signal indexed to the proton exchange product of HD was perceived on the Si-face but was not observed on the C-face. Moreover, in stark contrast to the H2 signal, the HD signal increased more abruptly on the Si-face, reaching a maximum within 10 s (Fig. 2f). Since photon-induced water splitting preferentially occurred at the 6H–SiC/water interface (Fig. S4c†), the pronounced HD signal, derived from interfacial HDO, indicated facile proton exchange and migration on the Si-face of 6H–SiC was exceedingly beneficial for overall water splitting.
The macroscopic water–SiC interaction was also studied by water contact angle measurement. The C-face was found to be hydrophobic with a large contact angle of 95.8°, consistent with the weak molecular water adsorption behavior on this face (Fig. 3a). Differently, the water contact angle on the Si-face was only 36.7°, showing that the interaction between water and the Si-face should be much stronger (Fig. 3b). This peculiar hydrophilic character of the Si-face could be explained by the facile proton exchange and migration, as well as the strong HB network constructed between the inner and the outer layer of water, which facilitated the overall water adsorption.14,26 Enlightened by the above experimental results, we further prepared Si- and C-face 6H–SiC photoanodes. A 200 nm thick Al layer was deposited on the backside of the highly conductive 6H–SiC films (0.8 cm × 0.8 cm) for making an ohmic contact for both the Si- and C-face 6H–SiC photoanodes, as evidenced by the linear I–V curve (Fig. 3c and S5†). A three-electrode photoelectrochemical cell was used to evaluate the PEC performances of the C- and Si-face 6H–SiC photoanodes in 0.5 M pH 6.8 Na2SO4 solution. Both 6H–SiC faces showed a low dark current in the linear sweep voltammetry scan, suggesting the absence of any chemical reactions in the dark. Under monochromatic light (λ = 410 nm, I0 = 30 mW cm−2), even though the C-face 6H–SiC photoanode exhibited a low onset-potential of 0.15 V versus the reversible hydrogen electrode (RHE) in the J–V curve, a remarkable current increase occurred only from 0.56 V, suggesting a typical sluggish water oxidation behavior due to the robust electron/hole recombination (Fig. 3d). As for the 6H–SiC photoanode with the Si-face, the onset potential of water oxidation is 0 V versus RHE even under neutral conditions, indicating that the OER can occur without external bias. Besides, a precipitous photocurrent increase occurred at as low as 0.10 V, which is a 0.46 V cathodic shift relative to the 6H–SiC photoanode with the C-face (Fig. 3d). Moreover, a plateau appeared around 0.40 V over the Si-face with a saturated photocurrent density of 1.01 mA cm−2, which is limited to the light intensity. It is noteworthy that to reach the plateau of photocurrent density, C-face 6H–SiC required a bias as large as 1.50 V.
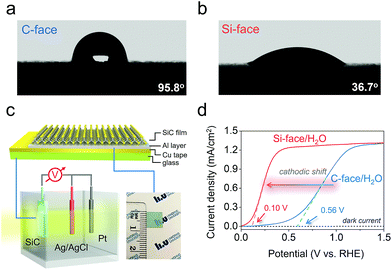 |
| | Fig. 3 Macroscopic water adsorption behavior and PEC performance of the C- and Si-face of 6H–SiC. Water contact angles of the (a) C- and (b) Si-face 6H–SiC. (c) Schematic illustration of the PEC cell and photoelectrode structure. (d) Current density–voltage (J–V) curves measured for the 6H–SiC photoanodes in 0.5 M pH 6.8 Na2SO4 solution under 410 nm monochromatic light. | |
2.3 Photoelectrochemical performance of SiC photoanodes
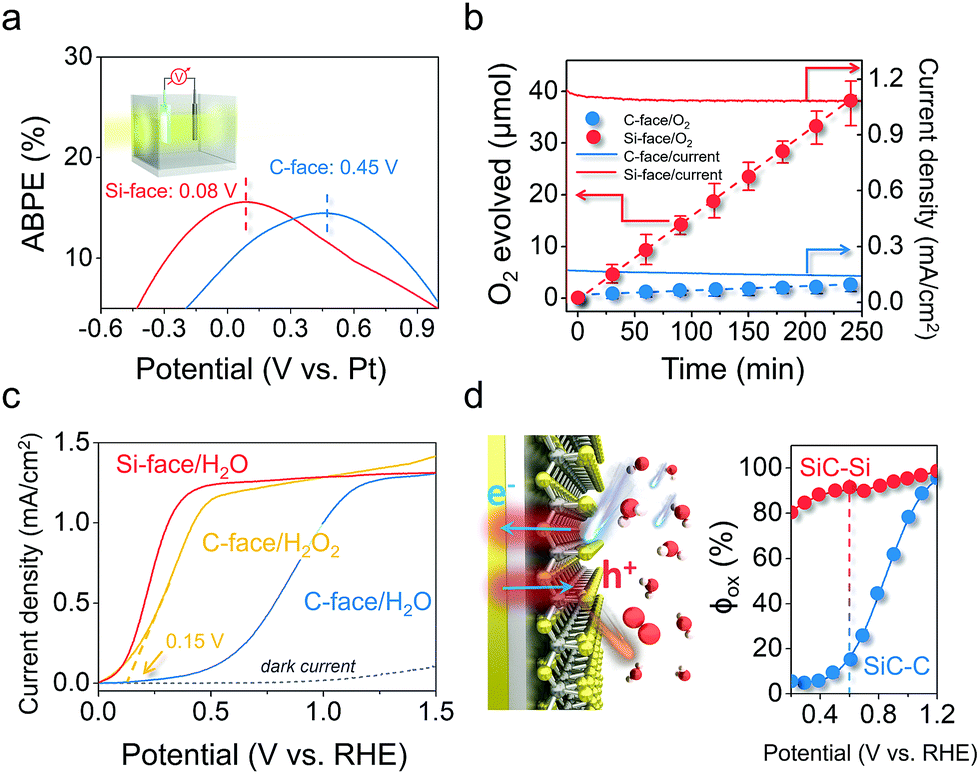
The remarkably reduced energy for triggering the OER on the Si-face could also be reflected in the applied bias photon-to-current efficiency (ABPE) in a two-electrode system. The maximum ABPE of 16.7% was achieved at an extremely low bias of 0.08 V on the Si-face under 410 nm monochromatic light, while it required a higher applied bias of 0.45 V to reach a comparable ABPE on the C-face (Fig. 4a). Correspondingly, the Si-face exhibited an obviously higher incident photo-to-current conversion efficiency than the C-face in the wavelength region of 300–410 nm (above the bandgap of 6H–SiC, 3.0 eV), even though they possessed almost the same light absorption capacity and identical optical band-edge absorption (Fig. S6†). Moreover, online O2 detection via gas chromatography (GC) confirmed that the photon-to-current conversion was mainly associated with stable O2 production with a current-to-O2 efficiency exceeding 90% under a bias of 0.4 VRHE in a three-electrode system (Fig. 4b). Since both the C- and Si-face 6H–SiC films possessed identical doping concentrations and comparable Fermi levels (Fig. S7†), we believe the onset potential, the photocurrent density and the evolved O2 differences over the photoanode with different faces are more related to surface charge kinetics rather than the flat band potential, conductivity, or bulk recombination. To prove this point of view, we quantitatively determined the quantum efficiency (φox) of water oxidation by surface holes according to the method developed by Dotan et al.33 The photocurrent density (J) arising from the OER is theoretically described as J = Jabsφsepφox, where Jabs is the theoretical photocurrent density of 6H–SiC assuming the absorbed photon conversion efficiency to be 100%, and φsep represents the charge separation yield of the photogenerated carriers (Fig. S8†). For an easily oxidized surrogate substrate like H2O2, surface recombination is completely suppressed with φox ≈ 1. Therefore, when probing water oxidation using the same photoelectrode, φox can be calculated from the ratio of J(H2O)/J(H2O2).34 To obtain J(H2O2), we performed linear sweep voltammetry over the photoanodes in the presence of 0.1 M H2O2 under the same conditions. Interestingly, the J–V curve of the 6H–SiC photoanode with the C-face performing H2O2 oxidation experienced a remarkable cathodic shift and sharp rise, resembling that of the 6H–SiC photoanode with the Si-face performing water oxidation (Fig. 4c). The calculated φox of the C-face in our case was very low at a bias below 0.6 V, reaching 80% at a higher bias of 1.0 V (Fig. 4d). As for the Si-face, it maintained a high φox approaching unity throughout the whole applied bias region (Fig. 4d). This demonstrated that separated holes on the Si-face more vividly participated in the surface water oxidation towards PEC oxygen evolution (Fig. 4d).
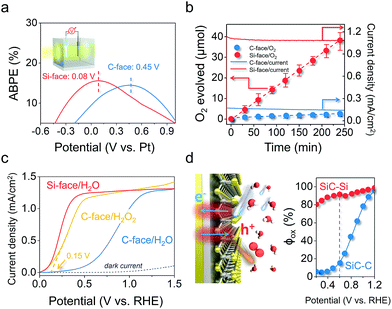 |
| | Fig. 4 Detailed PEC performance of the 6H–SiC photoanodes with Si or C-exposure. (a) ABPE of the 6H–SiC photoanodes in a two-electrode system using 0.5 M pH 6.8 Na2SO4 solution as the electrolyte. (b) Detected and calculated O2 production over the photoanodes at a bias of 0.4 VRHE in a three-electrode system. Solid lines show the corresponding J–t curves in 0.5 M pH 6.8 Na2SO4. Error bars represent standard deviation based on five independent experiments. (c) Comparison of J–V curves measured over the 6H–SiC photoanodes in 0.5 M pH 6.8 Na2SO4 with and without in 0.1 M H2O2. (d) The quantum efficiency (φox) for water oxidation by surface holes determined from the data in (c) for 6H–SiC photoanodes under PEC water oxidation conditions. Illumination: 410 nm monochromatic light. | |
2.4 Photoelectrochemical water oxidation kinetics on SiC photoanodes
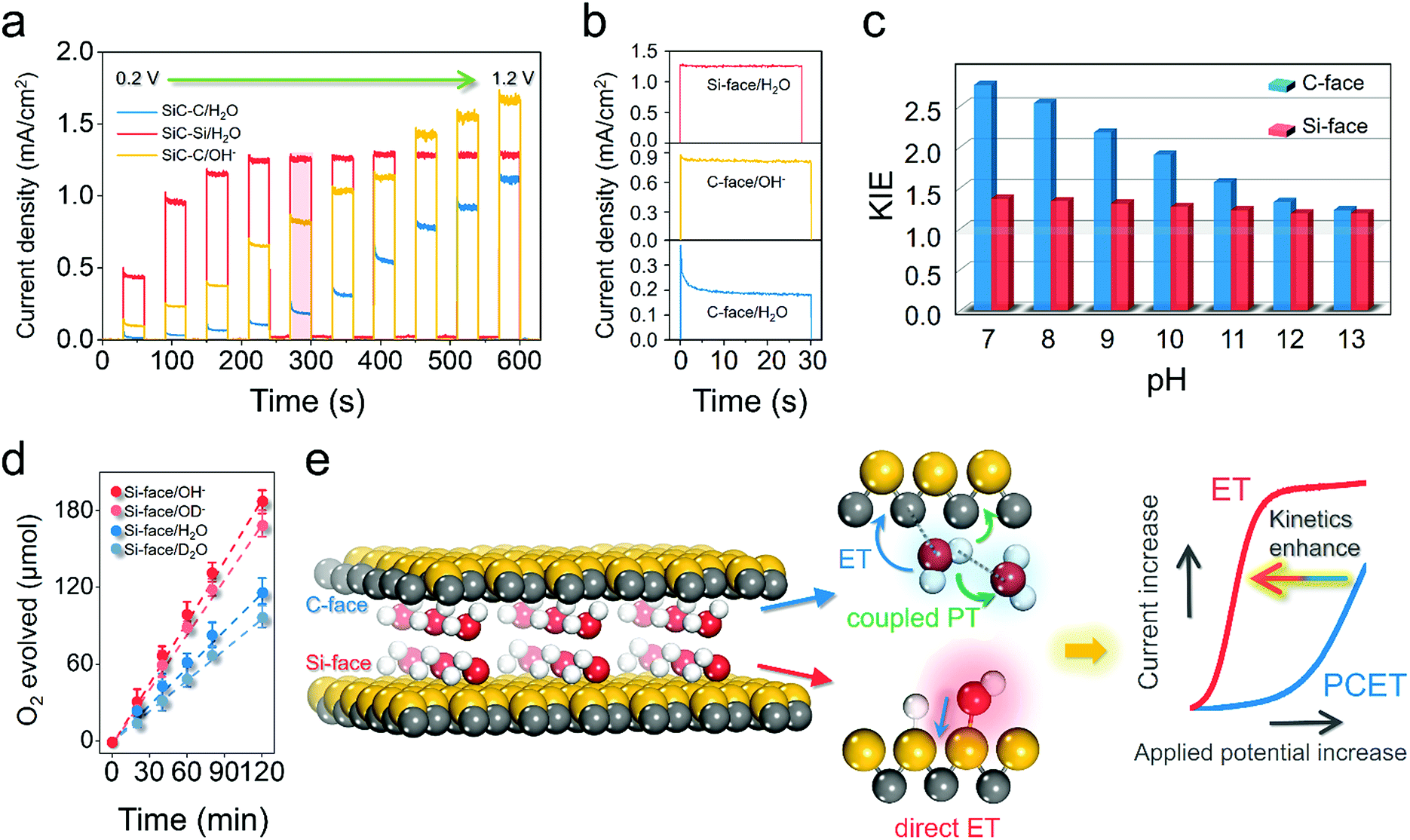
Apparently, the reason why the Si-face of 6H–SiC displayed lower charge recombination kinetics and stronger reaction kinetics for water oxidation than the C-face was closely related to its specific atomic structure. According to our theoretical and experimental results, the unique water dissociation mode on the Si-face of 6H–SiC with a facile proton exchange and migration character within the PT step was hypothesized to contribute substantially to its superior OER performance. To gain deep insight into the peculiar PT possibly occurring on the Si-face of the 6H–SiC photoanode, we first evaluated the surface charge transfer kinetics via monitoring the photocurrent transient change under a progressively increasing bias. The transient photocurrent of the C-face photoanode showed a large spike in the initial stage at either low or high applied potential, followed by a multiexponential decrease due to the charge carrier recombination (Fig. 5a).
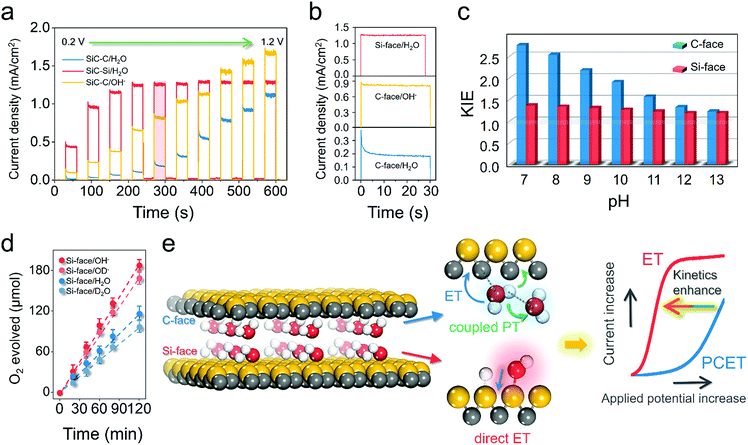 |
| | Fig. 5 Understanding the diverse mechanistic schemes of the photoelectrochemical OER on the 6H–SiC electrodes. (a) J–V curves measured over the photoanodes in 0.5 M Na2SO4 or 0.1 M NaOH at a progressively increasing applied bias. (b) Photocurrent transient decaying in (a) at an applied bias of 0.6 VRHE (highlighted in red). (c) The kinetic isotopic effect (KIE) in H2O and D2O with values calculated from the steady photocurrent ratio in H2O and D2O for photoanodes at various pH levels at an applied bias of 0.6 VRHE. (d) Detected O2 production over the photoanodes at a bias of 0.4 VRHE under neutral conditions (0.5 M Na2SO4 as the electrolyte) and alkaline conditions (0.1 M NaOH as the electrolyte). D2O and NaOD were used for H/D isotopic labelling. Error bars represent standard deviation based on five independent experiments. (e) Schematic illustration of water oxidation kinetics increase due to the different electron-proton transfer pathways on the C- and Si-face of 6H–SiC. Illumination: 410 nm monochromatic light. | |
According to the work by Zhong and Zhang et al., such photocurrent decaying shape represented a characteristic OER kinetic sluggishness due to the limited PT to water solvent or the catalytic surface during PCET steps.16,35 Typically, this major kinetic inadequacy could be circumvented in basic electrolytes, because OH− was easier to oxidize than H2O.35 As expected, the spikes were suppressed in 0.1 M NaOH, suggesting the suppression of surface recombination when kinetic PT was no longer involved (Fig. 5a and b). In contrast, the photocurrent response over the Si-face was not only substantially higher even under neutral conditions than the C-face, but also showed higher stability, as characterized by the small spikes at low bias and square shape of chopped photocurrent at high bias (Fig. 5b). This observation suggested that PT was no longer a kinetic bottleneck on the Si-face 6H–SiC photoanode in a neutral electrolyte. To acquire quantitative kinetic information on PT during the OER, the H/D isotopic labelling experiment was carried out. The steady-state photocurrent from the J–t curves after 120 s of irradiation was used to calculate the kinetic isotopic effect (KIE) in H2O and D2O (KIE = J(H2O)/J(D2O)) at different pH values. At a given moderate potential of 0.4 VRHE, pronounced KIE values over 2.0 were witnessed on the C-face at low pH (7–9) (Fig. 5c). These characteristic KIE values indicated that oxidation of water in the molecular state was the dominant reaction with O–H bond cleavage serving as the rate determining factor in the PCET steps of the OER. A similar mechanism was observed in biological and artificial molecular water oxidation systems.36,37 At higher pH (10–13), KIE values gradually decreased and became unity around 13, suggesting that the oxidation of OH− governed by ET was the major reaction. Differently, the Si-face photoanode exhibited a small KIE at either low pH or high pH, suggesting that the dominant step during the OER was ET. Moreover, we calculated the KIE values from the ratio of the evolved O2 gas under neutral or alkaline conditions (KIE = O2(H2O)/O2(D2O) or O2(NaOH)/O2(NaOD)). Consistently, the Si-face photoanode exhibited distinct low KIE values around 1.2 under either neutral (D2O) or alkaline (OD−, pH 12) electrolytes (Fig. 5d). Meanwhile, even under stronger near UV light irradiation (>80 mW cm−2), the Si-face photoanode was still able to maintain low KIE values towards the OER.38 This directly confirmed our theoretical prediction that on the Si-face of 6H–SiC, surface-catalyzed O–H breaking on water adsorption towards the formation of OH− was a barrierless process, which also promoted proton exchange and migration within PT during the OER, thus suppressing surface charge recombination in case of PCET and enhancing overall OER kinetics (Fig. 5e).
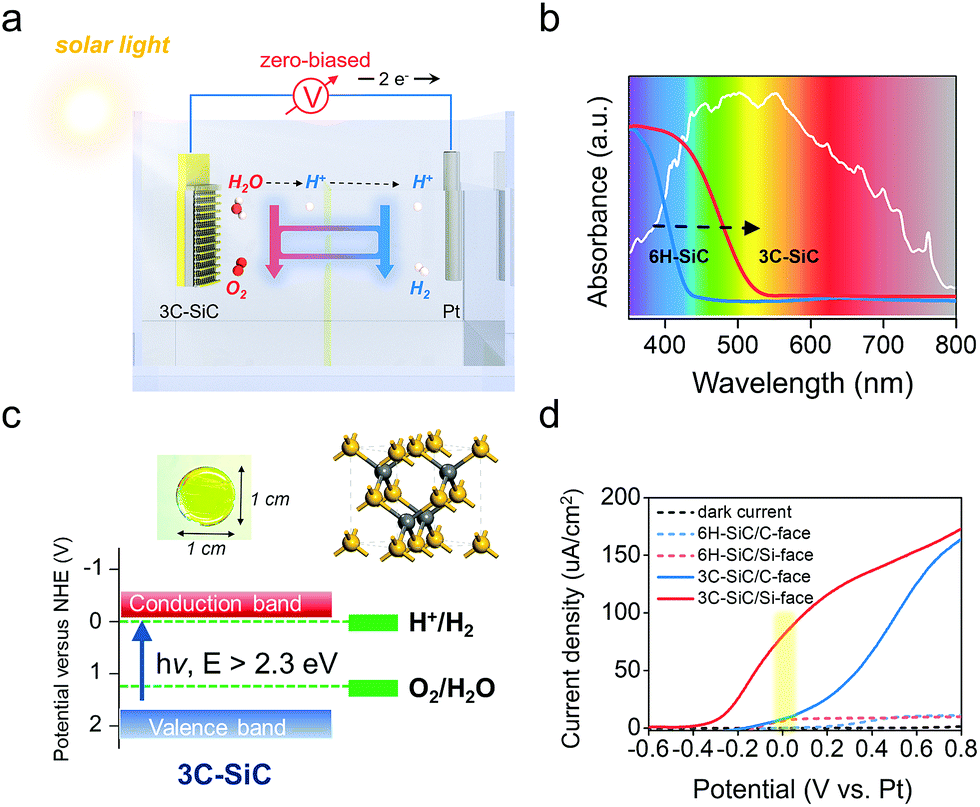
2.5 Construction of the zero-biased photoelectrochemical system
The ultimate goal for practical PEC device designing is to construct an operative two-electrode system for overall water splitting without any external bias (Fig. 6a), which is denoted as zero-biased water splitting.39–41 The 6H–SiC photoanode is a competitive material to enable such a conceptual design, because it can satisfy several key criteria, including high stability under strong acid or basic conditions and good transparency. Moreover, the Si-face of the 6H–SiC(0001) surface with manipulated PT behavior exhibited high OER kinetics even under neutral conditions, with its maximum of ABPE (0.08 V) achieved at a potential being close to zero in a two-electrode system (Fig. 4a). Unfortunately, 6H–SiC is a near-UV-light responsive semiconductor that largely limits its sufficient absorption to visible light. However, this proof-of-concept PT manipulation strategy we have explored over 6H–SiC can be perfectly applied to cubic 3C–SiC, a more ideal polymorph of SiC for PEC application due to its electronic structure.42,43 As compared with 6H–SiC, 3C–SiC possesses an extended absorption edge around 500 nm and a suitable bandgap around 2.3 eV that ensures ample solar light absorption, while simultaneously maintaining high potentials of the photogenerated charge carriers for water splitting reactions (Fig. 6b and c). Transparent yellowish single-crystalline 3C–SiC with the (111) polar surface exposed was grown via a high temperature sublimation epitaxial growth technique (Fig. 6c and S9†). Theoretical calculations demonstrated that water was preferentially dissociated on the Si-face of 3C–SiC (Fig. S10a and b†). In a two-electrode system (3C–SiC as the photoanode and Pt as the counter electrode), C-face exposed 3C–SiC displayed a remarkably high photocurrent response as compared with C-face 6H–SiC in 0.5 M pH 6.8 Na2SO4 solution under simulated solar light, while its ABPE reached a maximum only at 0.62 V (Fig. 6d and S10c†). Via preferentially exposing the Si-face, we found that the onset potential of 3C–SiC for the photoelectrochemical OER was remarkably shifted to −0.4 V, and the bias to achieve the maximum ABPE was reduced to 0.20 V in 0.5 M pH 6.8 Na2SO4 solution (Fig. 6d and S10c†). Under zero bias, an over 9 times photocurrent density increase was observed on the Si-face of 3C–SiC as compared with the C-face in the neutral pH electrolyte (Fig. S10d†). Noteworthy, in 1 M NaOH, where the sluggish PT was avoided, Si-face 3C–SiC could maintain the maximum of ABPE at zero bias in the two-electrode system, while the ABPE of C-face 3C–SiC experienced a remarkable cathodic shift to 0.19 V (Fig. S10e and f†). Optimizing the growth conditions to tune the doping and to enhance the crystalline quality with the aim of improving the photocurrent density is ongoing.
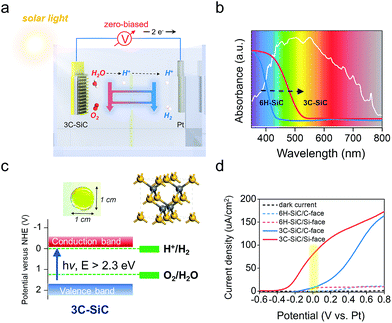 |
| | Fig. 6 Constructing a zero-biased two-electrode PEC system based on 3C–SiC. (a) Schematic illustration of the zero-biased two-electrode PEC cell for water splitting. (b) UV-vis absorption spectra of 6H– and 3C–SiC. The white line shows the emission spectra of the simulated solar light. (c) Color, crystal structure, and band structure of the as-prepared single-crystalline 3C–SiC film. (d) J–V curves measured for 6H–SiC and 3C–SiC photoanodes with both C- and Si-faces in 0.5 M pH 6.8 Na2SO4 under AM1.5G 100 mW cm−2 illumination. | |
3. Conclusions
In conclusion, using switchable polar SiC surfaces as the platform, we demonstrated that the bottleneck of water oxidation could be overcome via manipulating PT at the surface molecular level. On the C-face of SiC, where PCET governed the interfacial hole transfer for water oxidation, substantial energy loss was inevitable due to the PT steps. Via preferentially exposing the Si-face, we enabled surface-catalyzed barrier-less O–H breaking, with a facile proton exchange and migration character. This mechanistically shifted the rate limiting step of water oxidation from sluggish PCET on the C-face to a more energy-favorable ET on the Si-face. The understanding gained in this study unravels the vital role that PT plays in determining the kinetics of water oxidation and may open up new possibilities for the design of sophisticated photoelectrodes and zero-biased PEC systems via surface engineering.
4. Experimental section
4.1 Preparation of 6H–SiC(0001) films and photoanodes
The Si- and C-face 6H–SiC epilayers were grown by CVD on highly n-type doped Si- and C-face 4 degree off-axis 6H–SiC substrates, respectively. The thicknesses for both epilayers are around 30 μm. The Mott–Schottky measurements show an identical n-type doping concentration of 1.5 × 1015 cm−3 for both Si- and C-face 6H–SiC epilayers. Prior to the deposition of ohmic contacts on the backside of the films, they were chemically cleaned with acetone, ethanol, H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NH3:H2O2 (5:1:1), H2O:HCl:H2O2 (6:1:1), hydrofluoric acid (HF) and water. The samples were then fixed on a sample holder and placed inside a vacuum chamber together with an aluminum (Al) source for evaporation. The vacuum chamber was connected to a device calibrated for metal evaporation within the system such that desired thicknesses could be input and applied power for the evaporation is set automatically. The resulting contacts were made of a 200 nm layer of Al on the backsides of the samples to ensure good ohmic behaviors, as seen in Fig. S5.† The samples with deposited contacts on their backsides could be made into working electrodes by gluing them onto a piece of glass using copper tape. On some samples silver paste was used to further secure good contact between the copper and semiconductor. The whole electrode was then covered in epoxy resin, apart from the front surface of the semiconductor, which is supposed to be in contact with the electrolyte.
:NH3:H2O2 (5:1:1), H2O:HCl:H2O2 (6:1:1), hydrofluoric acid (HF) and water. The samples were then fixed on a sample holder and placed inside a vacuum chamber together with an aluminum (Al) source for evaporation. The vacuum chamber was connected to a device calibrated for metal evaporation within the system such that desired thicknesses could be input and applied power for the evaporation is set automatically. The resulting contacts were made of a 200 nm layer of Al on the backsides of the samples to ensure good ohmic behaviors, as seen in Fig. S5.† The samples with deposited contacts on their backsides could be made into working electrodes by gluing them onto a piece of glass using copper tape. On some samples silver paste was used to further secure good contact between the copper and semiconductor. The whole electrode was then covered in epoxy resin, apart from the front surface of the semiconductor, which is supposed to be in contact with the electrolyte.
4.2 Preparation of 3C–SiC(111) films
Bulk-like off-axis 3C–SiC(111) samples with a thickness of ∼1 mm were grown on 4 degree off-axis 4H–SiC substrates (SiCrystal) by the sublimation epitaxial growth technique.44,45 Then 300–400 μm thick free-standing 3C–SiC layers were obtained by polishing away the 4H–SiC substrates and the interfacial layer. To remove contaminants and oxide from the surface, the 3C–SiC substrates were chemically cleaned using acetone, ethanol, H2O:NH3:H2O2 (5:1:1), H2O:HCl:H2O2 (6:1:1), hydrofluoric acid (HF) and water. For making ohmic contact, 200 nm aluminum was deposited on the backside of each 3C–SiC sample by similar thermal evaporation.
4.3 Materials characterization
A Nicolet iS50FT-IR spectrometer was used to in situ record diffuse reflectance FTIR spectra on SiC. A contact angle goniometer (OCA-20, DataPhysics, Germany) was adopted to measure the water contact angles on SiC surfaces. The phase structure of SiC was studied by powder X-ray diffraction (XRD) on a Rigaku D/MAX-RB diffractometer. Photo-generated O2 and H2 were detected using a GC equipped with a mass spectrometer (Trace 1300-ISQ, Thermo). The surface topography of Si- and C-terminated samples was characterized with a 5 × 5 μm2 area using an atomic force microscope (AFM) in tapping mode. Electron-stimulated desorption (ESD) was carried out in an ultrahigh vacuum chamber (3 × 10−11 mbar), equipped with a low-energy electron gun (100 eV), a mass spectrometer and a molecular beam for water (H2O/D2O) deposition on the SiC substrate. Each individual scan on the SiC substrate was performed for 1 s, delivering a fluence around 3 × 1013 e− per cm2. For photon-simulated desorption (PSD) measurement, an Hg lamp with a 400 nm short-pass filter (>400 nm light was cut off) was used that did not remarkably influence the temperature of the SiC substrate.
4.4 PEC measurement
The PEC setup consisted of a cell with a quartz window on its side and a lid with several holder positions for electrodes. Unless otherwise stated, 0.5 M Na2SO4 solution of pH 6.8 was used as the electrolyte and 410 nm monochromatic light was used as the light source. Through the lid the electrodes were placed, the working electrode facing the quartz window, and the counter and reference electrodes close by. A Pt electrode was used as the counter and the Ag/AgCl was used as the reference electrode. J–V curves were measured using a scan rate of 10 mV s−1 and RHE was calculated using the Nernstian relation VRHE = VAg/AgCl + 0.0591 pH + 0.1976 V. Before the measurement, the electrolyte was purged with Ar for 0.5 h to remove the dissolved O2. For the isotopic labelling experiment, D2O was used as the solvent and NaOD was used to adjust the pH of the electrolyte. To determine the ABPE, a two-electrode system (working electrode and counter electrode) was used. The solar conversion efficiency of PEC water oxidation was also evaluated using ABPE in a strict two electrode system according to the following equation:
where Jph is the photocurrent density obtained under an applied bias Vb between the SiC electrode and the Pt counter electrode, 1.23 V is the standard water splitting reaction potential and Ptotal is the incident light intensity of 410 nm monochromatic light (30 mW cm−2).
4.5 Theoretical calculations
All the above calculations were performed using the first-principles density functional theory (DFT) + U calculations with the exchange–correlation energy functional, which were described by the generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation function.46 The projector augmented wave (PAW) method was applied to describe electron–ion interactions with a plane-wave cutoff energy of 400 eV.47,48 In our simulations, the (2 × 2) supercells with a vacuum thickness of 20 Å were built for Si- and C-terminated SiC surfaces, respectively. For optimization calculations, the energy and force converged to 10−5 eV per atom and 0.02 eV Å−1. A Monkhorst–Pack mesh was assigned as 5 × 3 × 1.49 Transition states were determined by the nudged elastic band (NEB) method.50,51 The interaction energy of the adsorbate water on the SiC surface was calculated as: ΔE = E(H2O/SiC) − E(SiC) − E(H2O). E(H2O/SiC) is the total energy of the water adsorbed SiC system. E(SiC) and E(H2O) are the energies of isolated SiC and H2O respectively. The charge density difference was calculated as: Δρ = ρ(H2O/SiC) − ρ(H2O) − ρ(SiC). ρ(H2O/SiC) is the density of the water adsorbed SiC system. ρ(H2O) and ρ(SiC) are the densities of the two isolated subsystems.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Swedish Research Council (Vetenskapsrådet, Grant No. 621-2014-5461), the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS, Grant No. 2016-00559), the Swedish Foundation for International Cooperation in Research and Higher Education (STINT, Grant No. CH2016-6722), the ÅForsk foundation (Grant No. 16-399 and 18-370), and Stiftelsen Olle Engkvist Byggmästare (Grant No. 189-0243).
Notes and references
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- K. Takanabe, ACS Catal., 2017, 7, 8006–8022 CrossRef CAS.
- C. Ding, J. Shi, Z. Wang and C. Li, ACS Catal., 2017, 7, 675–688 CrossRef CAS.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- H. Sheng, H. Zhang, W. Song, H. Ji, W. Ma, C. Chen and J. Zhao, Angew. Chem., Int. Ed., 2015, 54, 5905–5909 CrossRef CAS PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- X. Zhang and A. Bieberle-Hütter, ChemSusChem, 2016, 9, 1223–1242 CrossRef CAS PubMed.
- M.-T. Nguyen, S. Piccinin, N. Seriani and R. Gebauer, ACS Catal., 2015, 5, 715–721 CrossRef CAS.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. Chen, Y.-F. Li, P. Sit and A. Selloni, J. Am. Chem. Soc., 2013, 135, 18774–18777 CrossRef CAS PubMed.
- R. Nakamura and Y. Nakato, J. Am. Chem. Soc., 2004, 126, 1290–1298 CrossRef CAS PubMed.
- P. Salvador, J. Phys. Chem. C, 2007, 111, 17038–17043 CrossRef CAS.
- Y.-F. Li, Z.-P. Liu, L. Liu and W. Gao, J. Am. Chem. Soc., 2010, 132, 13008–13015 CrossRef CAS PubMed.
- W.-N. Zhao and Z.-P. Liu, Chem. Sci., 2014, 5, 2256 RSC.
- A. Valdés and G.-J. Kroes, J. Phys. Chem. C, 2010, 114, 1701–1708 CrossRef.
- Y. Zhang, H. Zhang, H. Ji, W. Ma, C. Chen and J. Zhao, J. Am. Chem. Soc., 2016, 138, 2705–2711 CrossRef CAS PubMed.
- Z. Huang, Y. Lin, X. Xiang, W. Rodríguez-Córdoba, K. J. McDonald, K. S. Hagen, K.-S. Choi, B. S. Brunschwig, D. G. Musaev, C. L. Hill, D. Wang and T. Lian, Energy Environ. Sci., 2012, 5, 8923 RSC.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef CAS PubMed.
- B. Iandolo and A. Hellman, Angew. Chem., Int. Ed., 2014, 53, 13404–13408 CrossRef CAS PubMed.
- D. H. van Dorp, N. Hijnen, M. Di Vece and J. J. Kelly, Angew. Chem., Int. Ed., 2009, 48, 6085–6088 CrossRef CAS PubMed.
- C.-H. Hao, X.-N. Guo, Y.-T. Pan, S. Chen, Z.-F. Jiao, H. Yang and X.-Y. Guo, J. Am. Chem. Soc., 2016, 138, 9361–9364 CrossRef CAS PubMed.
- G. Cicero, G. Galli and A. Catellani, J. Phys. Chem. B, 2004, 108, 16518–16524 CrossRef CAS.
- G. Cicero, A. Catellani and G. Galli, Phys. Rev. Lett., 2004, 93, 016102 CrossRef PubMed.
- F. Amy and Y. J. Chabal, J. Chem. Phys., 2003, 119, 6201–6209 CrossRef CAS.
- H. Sheng, H. Zhang, W. Song, H. Ji, W. Ma, C. Chen and J. Zhao, Angew. Chem., Int. Ed., 2015, 54, 5905–5909 CrossRef CAS PubMed.
- H. Li, J. Shang, H. Zhu, Z. Yang, Z. Ai and L. Zhang, ACS Catal., 2016, 6, 8276–8285 CrossRef CAS.
- H. Li, J. Li, Z. Ai, F. Jia and L. Zhang, Angew. Chem., Int. Ed., 2018, 57, 122–138 CrossRef CAS PubMed.
- T. Kimoto, A. Itoh and H. Matsunami, Appl. Phys. Lett., 1995, 66, 3645–3647 CrossRef CAS.
- C. D. Lane, N. G. Petrik, T. M. Orlando and G. A. Kimmel, J. Chem. Phys., 2007, 127, 224706 CrossRef PubMed.
- N. G. Petrik and G. A. Kimmel, J. Phys. Chem. C, 2009, 113, 4451–4460 CrossRef CAS.
- Z. Wu, W. Zhang, F. Xiong, Q. Yuan, Y. Jin, J. Yang and W. Huang, Phys. Chem. Chem. Phys., 2014, 16, 7051 RSC.
- Z. Zhang and J. T. Yates, Surf. Sci., 2016, 652, 195–199 CrossRef CAS.
- H. Dotan, K. Sivula, M. Grätzel, A. Rothschild and S. C. Warren, Energy Environ. Sci., 2011, 4, 958–964 RSC.
- D. K. Zhong, S. Choi and D. R. Gamelin, J. Am. Chem. Soc., 2011, 133, 18370–18377 CrossRef CAS PubMed.
- D. K. Zhong and D. R. Gamelin, J. Am. Chem. Soc., 2010, 132, 4202–4207 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed.
- T. Hisatomi, K. Takanabe and K. Domen, Catal. Lett., 2015, 145, 95–108 CrossRef CAS.
- D. Shao, Y. Cheng, J. He, D. Feng, L. Zheng, L. Zheng, X. Zhang, J. Xu, W. Wang, W. Wang, F. Lu, H. Dong, L. Li, H. Liu, R. Zheng and H. Liu, ACS Catal., 2017, 7, 5308–5315 CrossRef CAS.
- X. Wang, K.-Q. Peng, Y. Hu, F.-Q. Zhang, B. Hu, L. Li, M. Wang, X.-M. Meng and S.-T. Lee, Nano Lett., 2014, 14, 18–23 CrossRef CAS PubMed.
- Y. J. Jang, I. Jeong, J. Lee, J. Lee, M. J. Ko and J. S. Lee, ACS Nano, 2016, 10, 6980–6987 CrossRef CAS PubMed.
- Y. Liu, B. de Tymowski, F. Vigneron, I. Florea, O. Ersen, C. Meny, P. Nguyen, C. Pham, F. Luck and C. Pham-Huu, ACS Catal., 2013, 3, 393–404 CrossRef CAS.
- J. Wang, S. J. Xiong, X. L. Wu, T. H. Li and P. K. Chu, Nano Lett., 2010, 10, 1466–1471 CrossRef CAS PubMed.
- V. Jokubavicius, G. R. Yazdi, R. Liljedahl, I. G. Ivanov, J. Sun, X. Liu, P. Schuh, M. Wilhelm, P. Wellmann, R. Yakimova and M. Syväjärvi, Cryst. Growth Des., 2015, 15, 2940–2947 CrossRef CAS.
- V. Jokubavicius, G. R. Yazdi, R. Liljedahl, I. G. Ivanov, R. Yakimova and M. Syväjärvi, Cryst. Growth Des., 2014, 14, 6514–6520 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- N. A. Zarkevich and D. D. Johnson, J. Chem. Phys., 2015, 142, 024106 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta08631a |
|
| This journal is © The Royal Society of Chemistry 2018 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Jianwu
Sun
b and
Jianwu
Sun