
New insight into the structure of PuGaO3 from ab initio particle-swarm optimization methodology†
 a
a
Abstract
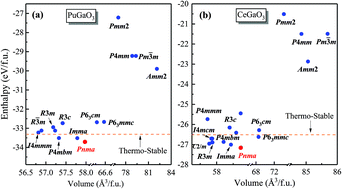
A systematic study of the phase stability, chemical bonding, and lattice dynamics properties of PuGaO3 is performed by means of unbiased particle-swarm optimization techniques combined with first-principles calculations. Phase stability analyses, including total energy, formation enthalpy, mechanical stability, and lattice dynamics, indicate that the ground-state structure of PuGaO3 at ambient pressure belongs to the space group Pnma. The fat-band dispersions and chemical bonding analysis suggest that Pnma PuGaO3 is an indirect band gap semiconductor (2.46 eV at the PBEsol + U level) at the Γ point and the Pu–O and Ga–O bonds in Pnma PuGaO3 are dominated by the ionic interactions. By examining the phonon spectra and phonon DOS, we provide a detailed insight into the lattice dynamics and thermodynamic properties of PuGaO3. In addition, the infrared absorption and Raman scattering spectra of PuGaO3 are predicted and further assigned at the center (Γ point) of the first Brillouin zone.
 Please wait while we load your content...
Please wait while we load your content...

