
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stoichiometry-dependent local instability in MAPbI3 perovskite materials and devices†
Vikas
Kumar
 *a,
Jérémy
Barbé
b,
Whitney L.
Schmidt
a,
Konstantinos
Tsevas
c,
Buse
Ozkan
a,
Christopher M.
Handley
a,
Colin L.
Freeman
a,
Derek C.
Sinclair
a,
Ian M.
Reaney
a,
Wing C.
Tsoi
b,
Alan
Dunbar
c and
Cornelia
Rodenburg
a
*a,
Jérémy
Barbé
b,
Whitney L.
Schmidt
a,
Konstantinos
Tsevas
c,
Buse
Ozkan
a,
Christopher M.
Handley
a,
Colin L.
Freeman
a,
Derek C.
Sinclair
a,
Ian M.
Reaney
a,
Wing C.
Tsoi
b,
Alan
Dunbar
c and
Cornelia
Rodenburg
a
aDepartment of Materials Science and Engineering, University of Sheffield, Mappin Street, Sheffield, S1 3JD, UK. E-mail: vikas.kumar@sheffield.ac.uk
b2SPECIFIC, College of Engineering, Swansea University, Bay Campus, Fabian Way, Swansea, SA1 8EN, UK
cDepartment of Chemical and Biological Engineering, University of Sheffield, Mappin Street, Sheffield S1 3JD, UK
First published on 12th November 2018
Abstract
Organometallic perovskite materials based on MAPbI3 achieve photovoltaic efficiencies as high as 22% for solar cells; however, the long-term stability of these perovskite materials is still a hurdle for applications. Here, we report the air and charge induced instabilities of MAPbI3 perovskite materials with different local stoichiometry using Secondary Electron Hyperspectral Imaging (SEHI). We find that individual grains which do not have the ideal ABX3 stoichiometry degrade faster than stoichiometric grains. We also observe that the degradation pathways depend on the local stoichiometry. Non-stoichiometric grains (with excess MAI) degrade from the centre of the grain and further degradation moves towards the grain boundary, whereas in stoichiometric grains degradation sets in at the grain boundary. We further use deliberately high doses of electron beam exposure to highlight the presence of local non-stoichiometry in device cross-sections of bias degraded devices which pinpoints different onset locations of degradation, depending on the local stoichiometry. These results, therefore, show that precise control of local stoichiometry is required to improve the stability of MAPbI3 and thus the lifetime of perovskite solar cells.
1. Introduction
Perovskite materials with an ABX3 crystal structure have unique chemical and physical properties which make them cost effective materials for future optoelectronic devices such as solar cells, light emitting diodes, lasers, detectors and transistors.1–6 So far, among all the combinations, MAPbI3 is one of the most studied perovskites for solar cell applications due to some of their best properties such as long diffusion length (1–175 μm), high optical absorption (∼105 cm−1) and mobility achieved by using different fabrication methods (e.g. spin coating, thermal evaporation, inkjet printing, doctor blade and drop-casting).7–13 However, the long-term stability of MAPbI3 has not been resolved and remains a major obstacle for its application and commercialisation.14 The degradation of MAPbI3 depends on several factors such as humidity, heat, trapped charge and UV light, but the degradation mechanism(s) and related time scales are still not clear.15–19 Such an understanding is hindered by the complexity arising from several competing processes taking place simultaneously, a strong influence of local stoichiometry on the degradation process, or a combination of both. The strong influence of the overall stoichiometry on degradation has been reported for complex (Cs and Br) containing perovskites as well as for simple MAPbI3 systems.20–22 However, none of these studies consider that the stoichiometry can vary locally23 (grain to grain) which might lead to different degradation processes within different grains. Using Secondary Electron Hyperspectral Imaging (SEHI), we followed the local degradation in response to different degradation triggers on the micron to nano-scale.We investigate environmental and induced-charge driven instability in MAPbI3 perovskites obtained by different processing methods. In particular, we compare different bulk MAPbI3 compositions to MAPbI3 thin films and device cross-sections, which were stored in air during the study. We also established that the top surface information is not a true representation of the whole device cross section when studying device stability because subsurface grains can exhibit different stoichiometry.24,25 Bulk MAPbI3 compositions were synthesized in powder form and then pressed into 6 mm diameter pellets with a thickness of 1.5 mm, resulting in materials with high crystallinity, which are anticipated as a future material for optoelectronic devices.26,27 We used MAPbI3 pellets with different starting compositions (e.g. stoichiometric, 5 mol% excess PbI2, and 5 mol% excess MAI) and studied their stability in air by SEHI. Clear differences were observed in local degradation pathways for the different starting compositions due to variation in the grain to grain stoichiometry. Variations in local degradation were also observed when charges were induced by the electron beam exposure or biasing of device structures. In the device structures, we present examples of MAPbI3 where the degradation is localised and triggered by the variation in the local stoichiometry between and within grains. Furthermore, the onset of induced-charge degradation can be observed to originate at different interfaces, also depending on local grain stoichiometry. The techniques described may also prove useful in studying the stability related to materials/phenomena such as organic–inorganic nano sheets for gas and energy storage, electronic devices, catalysis, drug delivery and any other hybrid materials in which local stoichiometry control is a fundamental requirement to warrant expected functionality.28
2. Experimental details
2.1 Synthesis and fabrication of pellets
SE spectra were collected for MAPbI3 pellets and thin films. Methylammonium iodide (MAI; GreatCell Solar) and lead iodide (PbI2; Sigma Aldrich, 99%) were mixed in an HDPE bottle with yttria-stabilized zirconia media (3 mm diameter) and ethanol (VWR Chemicals, ≥ 99.8%). The slurry was milled on a roller mill for 1 hour. The slurry was separated from the milling media and dried at 80 °C overnight to remove the solvent. The caked powder was hand ground in an agate mortar and pestle. The powder was stored in clear glass containers inside a desiccator, showing limited degradation of the bulk powder over time. For the stability study, MAPbI3 powder was pressed to form 6 mm diameter pellets with a thickness of ∼1.5 mm.2.2 Device fabrication for electrical bias-induced degradation of the perovskite
Anhydrous dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF), anhydrous ethanol, anhydrous chlorobenzene, anhydrous ethanolamine, anhydrous 2-methoxyethanol, bathocuproine (BCP, 96%), nickel acetate (NiAc, 98%) and lead iodide (PbI2, 99%) were purchased from Sigma-Aldrich. Methylammonium iodide (MAI) was purchased from Dyesol. The electron transport material [6,6]-phenyl-C61 butyric acid methyl ester (PCBM) was obtained from Solenne BV. To prepare the NiOx solution, a 0.2 M solution of nickel acetate tetrahydrate was dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.012 volume ratio of 2-methoxyethanol:ethanolamine and stirred for 1 h at 60 °C. The MAPbI3 precursor solution was prepared by mixing MAI and PbI2 (1:1 and 1:1.05 molar ratios) in DMF/DMSO (4:1 volume ratio) with a concentration of 804 mg ml−1 at 60 °C for 2 h. PCBM was dissolved in chlorobenzene at a concentration of 20 mg ml−1 and stirred overnight at 60 °C. BCP was dissolved in anhydrous ethanol at a concentration of 0.5 mg ml−1. The NiOx layer was deposited via spin-coating on previously cleaned 15 Ω □−1 ITO/glass substrates (Lumtec). Subsequent layers were prepared under nitrogen in a glove box. The perovskite precursor solution was spin coated using the chlorobenzene anti-solvent dripping technique to promote crystallization followed by annealing at 100 °C for 10 min. After cooling, the PCBM solution was spin-coated onto the MAPbI3 layer at 4000 rpm for 30 s, followed by spin-coating the BCP solution at 6000 rpm for 20 s. Finally, 100 nm thick silver counter electrodes were evaporated at 10−1 torr using an Edwards 306 thermal evaporator, forming devices with an active area of 0.15 cm2.
:0.012 volume ratio of 2-methoxyethanol:ethanolamine and stirred for 1 h at 60 °C. The MAPbI3 precursor solution was prepared by mixing MAI and PbI2 (1:1 and 1:1.05 molar ratios) in DMF/DMSO (4:1 volume ratio) with a concentration of 804 mg ml−1 at 60 °C for 2 h. PCBM was dissolved in chlorobenzene at a concentration of 20 mg ml−1 and stirred overnight at 60 °C. BCP was dissolved in anhydrous ethanol at a concentration of 0.5 mg ml−1. The NiOx layer was deposited via spin-coating on previously cleaned 15 Ω □−1 ITO/glass substrates (Lumtec). Subsequent layers were prepared under nitrogen in a glove box. The perovskite precursor solution was spin coated using the chlorobenzene anti-solvent dripping technique to promote crystallization followed by annealing at 100 °C for 10 min. After cooling, the PCBM solution was spin-coated onto the MAPbI3 layer at 4000 rpm for 30 s, followed by spin-coating the BCP solution at 6000 rpm for 20 s. Finally, 100 nm thick silver counter electrodes were evaporated at 10−1 torr using an Edwards 306 thermal evaporator, forming devices with an active area of 0.15 cm2.
2.3 Hyperspectral imaging
The basic principles of the experimental method for hyperspectral imaging have been reported elsewhere.23 In this work, an FEI Helios NanoLab G3 UC SEM was employed. All samples were imaged using a through-lens detector (TLD) at a working distance of 4.1 mm with a beam current of 13 pA and an accelerating voltage of 1 kV. The dwell time was adjusted to 50 ns for all SEHI images. Hyperspectral imaging window selection was performed by varying the bias to the mirror electrode.29 SE image stacks were taken by increasing the mirror voltage (−1.0 to 8 eV), with a step size of 0.2 eV.2.4 Computational simulation
Atomic simulations were performed using the GULP code with a force field designed within the group.30,31 Energy minimisations were carried out on 4 × 4 × 4 bulk cells with an effective formula of MA32Pb32I96. Defects in the material were analysed using the Mott–Littleton method to examine isolated MA, Pb and I defects along with charge neutral defect clusters (MAI and PbI2). The convergence of defect energies was confirmed with cut-offs of 16 Å and 30 Å for regions 1 and 2A, respectively. Further absolute concentration defects were considered by using the supercell method and eliminating the Schottky defects of MAI and PbI2 in concentrations of 0.03, 0.06 and 0.09 (relating to creating 1, 2 and 3 defects in the cell). Multiple different cluster structures were calculated and the lowest energy defects are reported. The solution energy (Esol) for each defect was calculated by generating a set of reaction schemes such as| VPbI2: MA32Pb32I96 = MA32Pb31I94 + PbI2 |
| Esol (VPbI2): E(MA32Pb31I94) + E(PbI2) − E(MA32Pb32I96), |
3. Results and discussion
3.1 Effect of stoichiometry on MAPbI3 stability in air
To study the effect of stoichiometry on perovskite stability we used three different starting compositions of bulk MAPbI3 (pellets) which were stoichiometric, 5 mol% overall excess PbI2, and 5 mol% overall excess MAI. During the experiments all the pellets were stored in air in the dark with a relative humidity of ∼40%. Fig. 1 shows the standard surface morphological images of these pellets which were collected for fresh pellets (as received) and after three and six weeks' storage in air, respectively. Compared to the as-received pellets all the aged (after three and six weeks' storage) MAPbI3 pellets had degraded and show the formation of new phases (brighter contrast in LVSEM images). The surface morphological images of a stoichiometric MAPbI3 pellet before and after degradation are shown in Fig. 1a and c. The as-prepared stoichiometric MAPbI3 shows no sign of degradation, whereas aged MAPbI3 (Fig. 1b) after 3 weeks exhibits some initial degradation emerging at grain boundaries (indicated by red arrows and in the high magnification inset image) and extending towards the centre of the grain. This process continues until the grains completely degrade which is more clearly observed in Fig. 1c collected after 6 weeks. The progression of degradation due to air exposure of stoichiometric MAPbI3 seen here is similar to the degradation pattern for MAPbI3 thin films under moisture conditions recently observed by Wang et al.16 Here, we have evidence that the stoichiometry of MAPbI3 perovskite materials affects the degradation process, because under identical storage conditions the pellets with excess PbI2 degrade faster (Fig. 1e) than the pellets with excess MAI (Fig. 1h) or stoichiometric MAPbI3 (Fig. 1b). Compared to stoichiometric MAPbI3, where degradation only affects the edges of grains, a dense coverage of bright nano structures appears on the surface of the pellets with excess PbI2. We note that this is the case for the majority of the grains when aged (three weeks in air). This shows the fast degradation of excess PbI2 materials but the pattern remains similar to that of stoichiometric MAPbI3 in terms of degradation onset occurring from the grain boundaries. This is clearly observed in completely degraded materials (Fig. 1c and f) collected after 6 weeks. Hence, we propose that the presence of excess PbI2 promotes the degradation of these perovskite materials. Furthermore, we observe that the surface morphologies of the almost completely degraded stoichiometric MAPbI3 pellet and the pellet with excess PbI2 (Fig. 1c and f) are different. | ||
| Fig. 1 Standard LVSEM surface morphological images of MAPbI3 pellets at an electron beam voltage of 1 kV and a beam current of 13 pA; the (a) as-prepared stoichiometric MAPbI3 pellets; (b) degraded stoichiometric pellets after 3 weeks; (c) completely degraded stoichiometric pellets after 6 weeks; (d) as-prepared excess PbI2 pellets; (e) degraded excess PbI2 pellets after 3 weeks; (f) completely degraded excess PbI2 pellets after 6 weeks; (g) as-prepared excess MAI pellets; (h) degraded excess MAI pellets after 3 weeks; (i) completely degraded excess MAI pellets after 6 weeks. | ||
In 6 weeks aged excess PbI2 pellets (Fig. 1f) we observe a very high prevalence of sheet or plate like structures (indicated by the red arrow in Fig. 1f) compared to the 6 weeks aged stoichiometric MAPbI3 in Fig. 1c. This is more clearly seen at higher magnification (see ESI Fig. S1†) and occurs in most but not all grains in the excess PbI2 pellets. We suspect the latter might be due to local variations in the stoichiometry (composition variations) between the initial perovskite grains. To map such local variations before and after the degradation we have used SEHI as discussed further below. Fig. 1g–i reveal the surface morphological images of excess MAI pellets before and after degradation. These show a very different degradation path to that for stoichiometric MAPbI3 and excess PbI2 pellets. Excess MAI pellets exhibit localised degradation that first emerges at the centre of the grain (indicated by black arrows in Fig. 1h). Then the degradation front moves towards the grain boundary. The degraded regions in the excess MAI pellet appear bright and become porous after degradation (see ESI Fig. S2†). We attribute the bright porous appearance of the degraded grains to the decomposition of MAPbI3 into a volatile phase, as reported by Han et al.15 We also noted the non-uniform manner in which individual grains degrade at very different rates compared to their neighbouring grains (see ESI Fig. S3†). The latter behaviour suggests that it is not true that degradation always originates at grain boundaries. Whether this is true depends on the stoichiometry of the individual grains as some small modifications in local stoichiometry have a strong impact on the degradation pattern of individual grains. For instance, in Fig. 1i, dark grey areas are observed (indicated by the blue arrow) that represent regions with excess MAI as determined by their relative contrast in SEHI at low energies (see ESI Fig. S4†). The absence of peaks in the X-ray diffraction data for MAI or MAI rich phases (not shown here) suggests that they are amorphous. Such areas only occur in pellets with excess MAI and do not show any obvious signs of degradation as a result of air exposure in the Secondary Electron (SE) images or SE spectra. The experimental observation of differences in the rate of degradation for the above three systems can be understood in terms of the energy required to create a defect that can be modelled using the Mott–Littleton method. Fig. 2 shows the defect energies of MAI and PbI2. We can immediately see that the MAI vacancies are energetically favourable (negative energies) and therefore would be expected to spontaneously occur. This compares with the PbI2 defects which are energetically unfavourable although the solution energies are still relatively low. Hence we can assume that the variation in defect energies is responsible for the experimentally observed differences in the degradation rate and paths seen in Fig. 1a–i. The second interesting feature is the variation of the energies as the concentration of the defects is increased. For PbI2 the solution energy increases with each subsequent defect; the loss of the first defect takes only 1.5 eV but to remove a subsequent PbI2 then takes 2.1 eV – a total of 0.6 eV more energy and this trend continues as more PbI2 is removed. For MAI, however, we see the opposite trend. The first MAI releases an energy of 0.11 eV but the subsequent MAI releases a further 0.19 eV (total energy −0.30 eV). This suggests that each subsequent loss of a MAI defect is more favourable. Therefore, the further the material is from stoichiometry with excess PbI2 (i.e. less MAI in relation to PbI2), the more readily further losses will occur. This agrees closely with the results observed in Fig. 1 where material with excess PbI2 degrades the fastest (Fig. 1e). In contrast, stoichiometric and excess PbI2 pellets degrade from the grain boundary inwards because the only pre-existing defects are those found on the grain boundaries. Next we investigate if these very different degradation paths lead to different degradation products. To test this, we resort to SEHI.
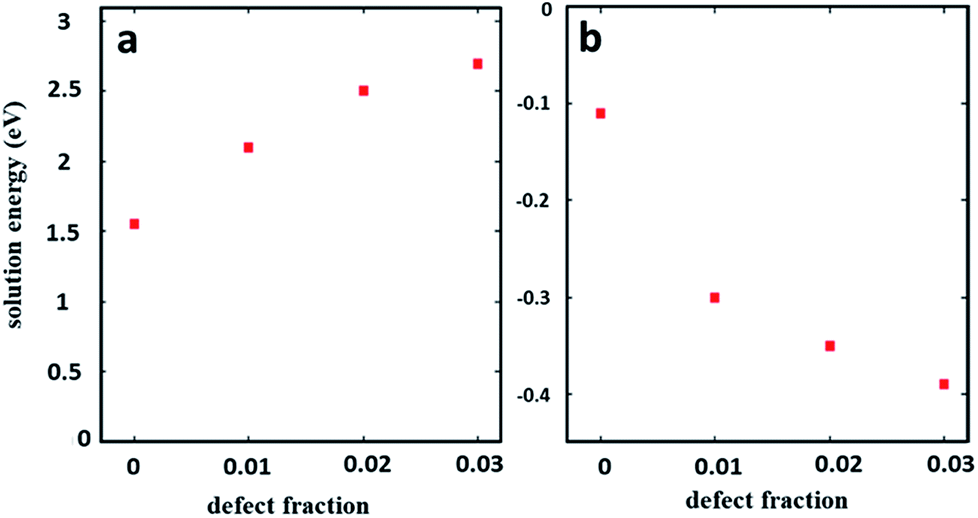 | ||
| Fig. 2 Solution energies for (a) PbI2 and (b) MAI vacancies. A defect concentration of 0 refers to an infinitely dilute defect calculated via the Mott–Littleton method. | ||
3.2 Mapping of local degradation products via SE hyperspectral imaging (SEHI)
First we identify regions within the SE emission spectra that show strong intensity changes when comparing spectra taken from the overall stoichiometric perovskites to those taken from perovskites with excess material added (e.g. 5 mol% excess PbI2 and 5 mol% excess MAI) (see Fig. 3a) to the nominal starting composition. The SE spectrum of pure PbI2 pellets is also plotted in Fig. 3a for comparison. The most intense peaks of the stoichiometric and excess MAI pellets completely overlap, suggesting that the addition of excess MAI has no major impact on this peak. Further, an additional peak (∼3.50 eV) appears for pellets with excess MAI which is absent for stoichiometric MAPbI3 and pellets with excess PbI2. Therefore, we suggest that this new peak (at 3.5 eV) is due to the presence of excess MAI/MAI-rich perovskite and thus can be used to map amorphous MAI-rich regions as long as we can confidently describe its chemical origin. To confirm that this is a MAI peak, we collected the SEHI images of excess MAI pellets using energy windows at 3.5 and 10 eV, respectively. MAI regions are expected to appear bright only in images taken with the 3.5 eV energy window – which is the case (see ESI Fig. S4†). They appear dark in images collected using the energy window of 10 eV. This means that MAI-rich regions emit low energy SEs that cannot be detected selectively in standard SEM. This is in good agreement with our previous work, where we observed similar behaviour for bromide-based perovskite materials.23 The SE spectra of excess PbI2 pellets are also represented in Fig. 3a. Here the position of the most intense peak is different to that obtained from stoichiometric MAPbI3 and pellets with excess MAI but overlaps with that of pure PbI2. Thus the spectrum clearly reflects the presence of excess PbI2 (originating from the areas indicated by the red and black dotted arrows on the surface of as well as in between perovskite grains in Fig. 1d). This observation confirms that SEHI can be used as a novel tool to probe local stoichiometry on the nano-scale provided the correct energy windows (indicated in Fig. 3a) are selected for imaging as follows: (1) the 1.4–2.0 eV window representing either MAI (MAI-rich perovskite) or the MAPbI3 perovskite as the peaks are overlapping; (2) the 2.7–3.3 eV window representing pure PbI2; (3) the 3.3–3.6 eV window to map the presence of excess MAI (or MAI-rich perovskite). Additional SE peaks observed at higher energy (above 4.0 eV) are less reliable for chemical mapping because material topography can dominate the overall contrast in this energy range.23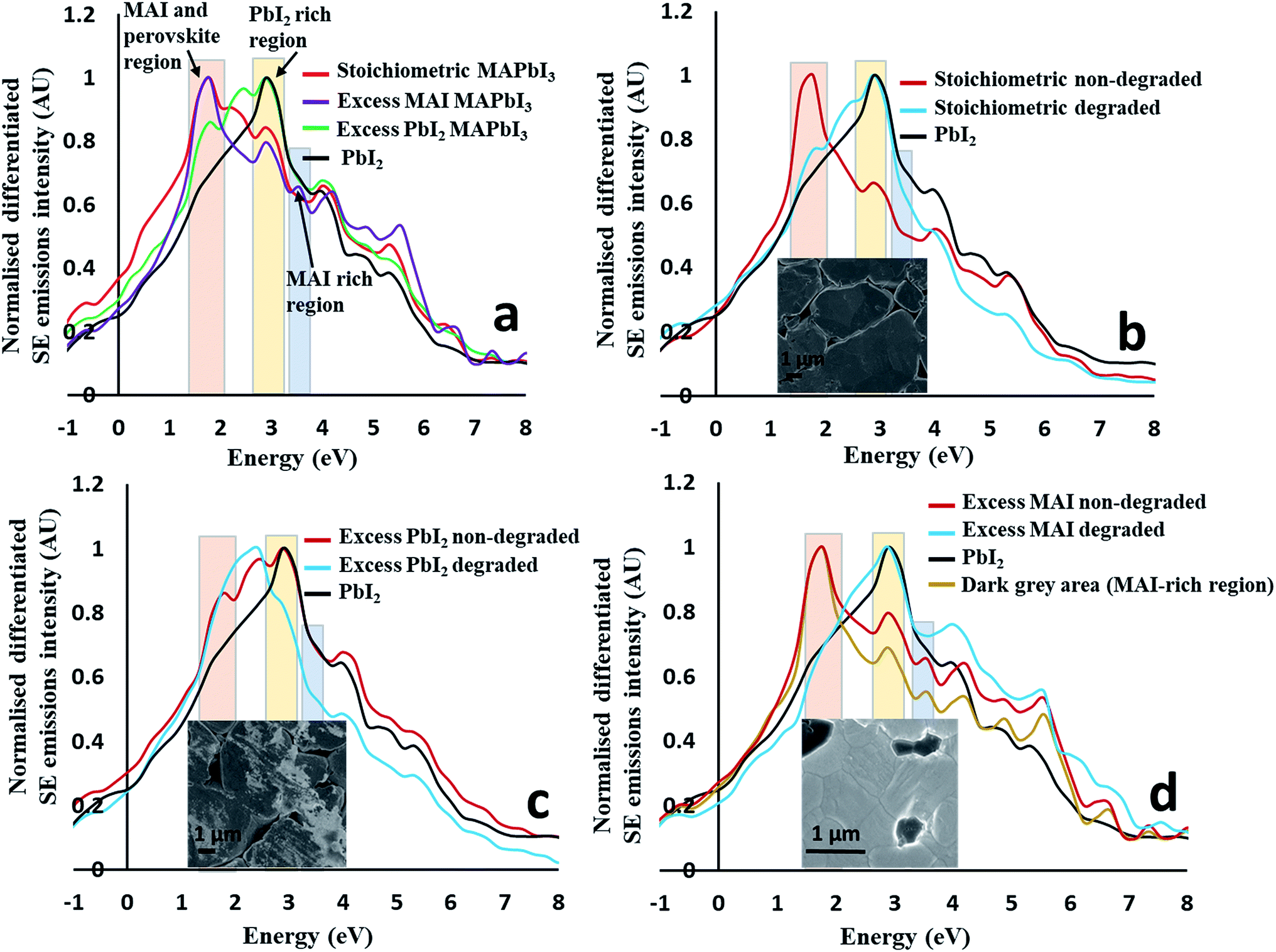 | ||
| Fig. 3 Differentiated SE spectra of (a) the as-prepared stoichiometric MAPbI3, excess PbI2 and MAI pellets and PbI2; (b) the as-prepared and degraded stoichiometric MAPbI3 and PbI2; (c) the as-prepared and degraded excess PbI2 pellets and PbI2; (d) the as-prepared and degraded excess MAI pellets and PbI2. | ||
To identify degradation products, we collected the spectra shown in Fig. 3b–d that contain the differentiated SE spectra of stoichiometric (Fig. 3b), excess PbI2 and excess MAI (Fig. 3c and d) pellets before and after degradation. For all pellet compositions, a change in the SE spectra was observed as a result of degradation. After the degradation of stoichiometric MAPbI3 (Fig. 3b), the most intense peak shifted from the first energy window (1.4–2.0 eV) to the second (2.7–3.3 eV), hence completely overlapping with the pure PbI2 region. This suggests that stoichiometric MAPbI3 completely decomposes to PbI2 and a volatile component after degradation (as seen in Fig. 1c).
The degradation of the pellet with excess PbI2 in air (Fig. 3c) is quite different, where the most intense SE peak of the degraded perovskite material is in a different position (2.4 eV) to that of pure PbI2 and not associated with any of the energy windows defined in Fig. 3a. Pellets with excess PbI2 perovskites therefore decompose to some other phase(s). This is consistent with the difference in morphology between degraded stoichiometric pellets and degraded excess PbI2 pellets (for more images see ESI Fig. S1†). The degradation mechanism of pellets with excess MAI in air (Fig. 3d) is similar to that of the stoichiometric MAPbI3 (Fig. 3b), because the most intense peak is again shifted from the first to the second energy window (similar to stoichiometric MAPbI3). For both (stoichiometric and excess MAI) pellets the 1.4–2.0 peak is missing suggesting the complete degradation of the perovskite to pure PbI2 and potentially another phase(s) as some high energy peaks (above 4.0 eV) show higher intensity (Fig. 3d) after the degradation of the pellet with initial excess MAI. However, we find that these peaks might not be reliable because for this energy region the SE emissions due to topography can influence the spectrum.23 Based on the above investigations, we conclude that the degradation in air strongly depends on the stoichiometry of the perovskite material which can vary locally (grain to grain). The progress of local degradation can best be observed by collecting images from two energy regions, a window centred around 3.5 eV (to monitor the organic component that cannot be detected efficiently by EDX) and a second window centred around 3.0 eV to observe the PbI2. Furthermore, the observed near final degradation pattern (inward or outward growth of the degradation front) is an indication of the initial local stoichiometry and might be used to adjust the processing conditions.
3.3 Charge (electron beam) induced instability of MAPbI3
We further analysed the electron beam induced instability for the various initial pellet compositions using an accelerating voltage of 1 kV and a beam current of 13 pA. There are reports suggesting that an electron beam can damage the perovskite surface by defect formation and phase transformation.32 To check the dependence of electron beam damage on the initial stoichiometry, we analysed the as-prepared excess MAI pellets and stoichiometric MAPbI3 pellets under identical electron beam conditions. Fig. 4 shows the SEM surface morphologies of the above compositions under repeated electron beam exposure of the same area. Fig. 4a–c are the first, second and third scan surface morphological images of a pellet with initial excess MAI, whereas Fig. 4d–f are the corresponding images for stoichiometric MAPbI3. Fig. 4b shows that a 1 kV electron beam can promote the decomposition of a perovskite surface when it initially contains some excess MAI. It generates localised structural defects as revealed on the surface during the second scan with an electron dose of ∼1.25 × 1016 el− cm−2 (equivalent to ∼2000 mW cm−2 or 20 Suns at AM 1.5 G) that grow further during the third scan. The growth of these very local bright nano-structured regions is due to the electron beam induced decomposition of the perovskite into PbI2 and gaseous species (CH3NH2 and HI),33,34 as 90% of the beam energy (as estimated from the CASINO Monte Carlo program, see Fig. S5 and S6 in the ESI†) is dissipated within the top 2.5–3.0 nm. This promotes decomposition while enabling the release of gaseous products. Due to the high vacuum environment in the SEM chamber the gaseous species are removed instantly, leaving the bright nano-structured regions which we attribute to PbI2. Similar to the degradation in air, the charge induced degradation of perovskites with initial excess MAI is also localised suggesting that individual grains decompose due to the variation of their initial non-stoichiometry. The perovskite grains (non-stoichiometric with excess MAI) most likely contain a greater concentration of point defects from which MAI degrades fast compared to the stoichiometric grains. Therefore, all of these defects appear to be related to the initial chemical and structural quality of the individual grains. Due to the non-uniform stoichiometry, the grain boundaries in excess MAI perovskites are also unstable (see ESI Fig. S6†) under charge exposure and frequently appear cracked (indicated by the black arrow in Fig. 4b and ESI Fig. S7†). In contrast, for overall stoichiometric perovskites (Fig. 4d–f), electron exposure at 1 keV has no impact on the material surface for identical amounts of electron dose exposure as in Fig. 4a–c. Based on the above results, we propose that prolonged scanning with a 1 keV electron beam is an effective tool to reveal the presence of localized non-stoichiometry if SEHI is not available.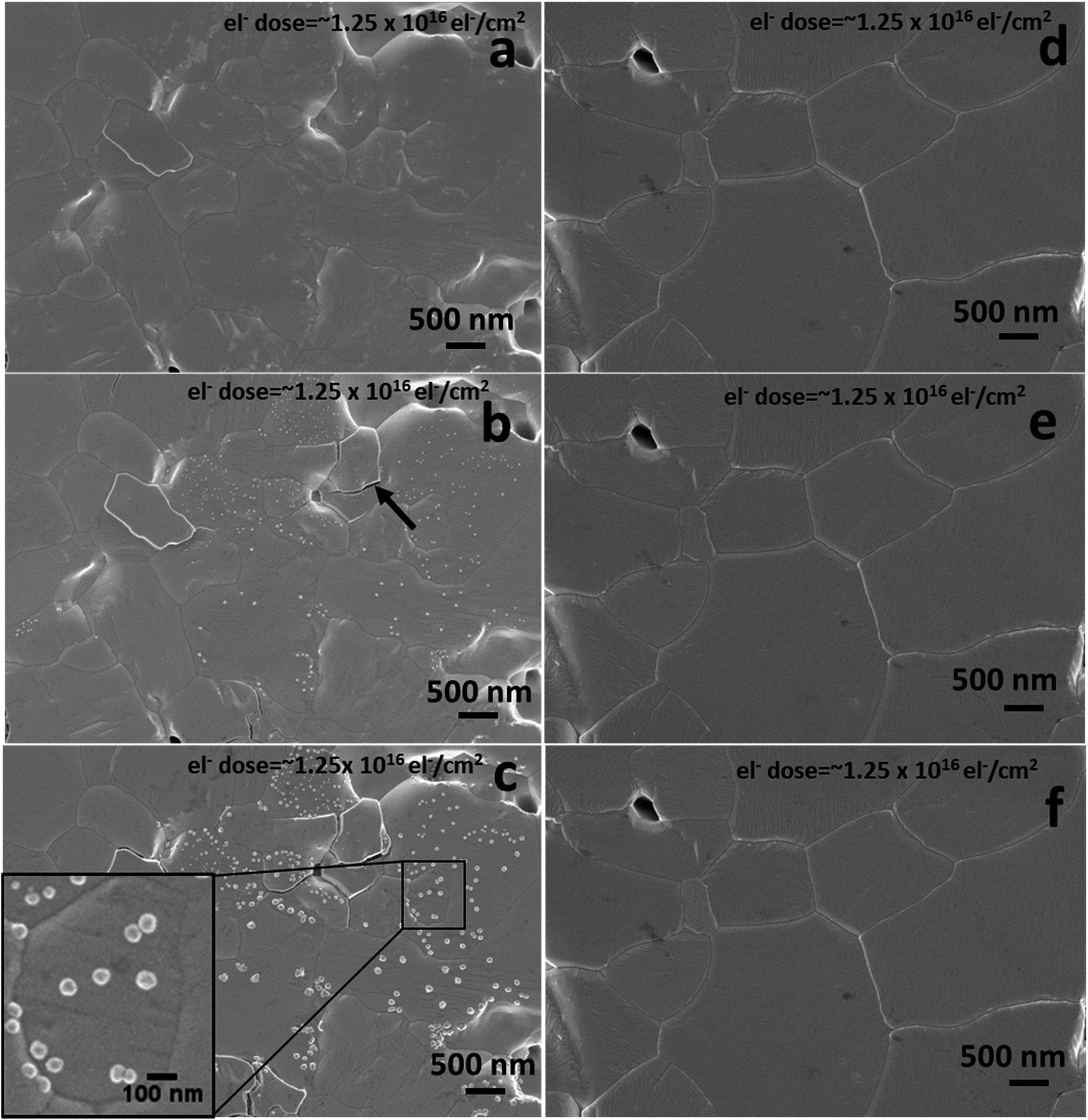 | ||
| Fig. 4 Standard LVSEM surface morphological images of MAPbI3. (a–c) The as-prepared excess MAI pellets for the first, second and third scan; (d–f) stoichiometric MAPbI3 pellets for the first, second and third scan, respectively. | ||
3.4 Comparison of MAPbI3 pellets and thin films
To understand the impact of MAPbI3 morphology on the SE spectra, we first collected the SE spectra of stoichiometric MAPbI3 pellets and thin films under identical operational conditions. The surface morphologies of the pellets and thin films are very different with the grain sizes in the pellets and the thin films typically being ∼10 μm and 300–400 nm, respectively (see ESI Fig. S8†). The SE spectra of both the perovskite materials under identical electron beam conditions in the energy range of −1 to 8.0 eV are shown in ESI Fig. S9.† The most intense peaks for both the pellets and the thin films fell perfectly within the first energy window (1.4–2.0 eV in Fig. 3a) and the features at 3.5 eV were not visible here, which means both the bulk and thin film perovskite materials are stoichiometric and that the synthesis method has little impact on the SE spectra of MAPbI3 in the lower energy range, even though the surface morphologies of the pellets and thin films are different (ESI Fig. S8†). Hence, we can apply the findings of the above investigations on pellets to the case of thin films in device cross-sections to map their degradation in order to elucidate degradation mechanisms.3.5 Charge (bias) induced instability of MAPbI3 devices
Stoichiometric and non-stoichiometric (with 5% excess PbI2) MAPbI3 solar cell structures were examined. Note that the tested solar cell devices were degraded using a forward bias voltage of 1 V for 1.5 h in ∼40% RH air in the dark.35 The J–V (current–voltage) characteristics showed a dramatic change in performance for both the compositions after the application of an electrical bias. The stoichiometric device lost ∼45% of its initial power conversion efficiency (PCE) after bias for 1.5 h while the non-stoichiometric device lost ∼60% of its initial PCE (see ESI Fig. S10 and Table S1†). This suggests that the devices containing 5% excess PbI2 (non-stoichiometric) are more unstable under applied bias, which is similar to what was discussed in Fig. 1b and e for stoichiometric and PbI2-rich pellets. This can further be explained by LVSEM inspection of device cross-sections before (Fig. 5a and c) and after (Fig. 5b and d) bias induced degradation. Electrical bias degraded devices (Fig. 5b and d) exhibit a similar localised degradation (indicated by the black circle) to that of prolonged electron beam exposure, where some grains degrade quickly whereas neighbouring grains remain unaffected. Additionally, the electron beam exposure triggered the accelerated growth of bright PbI2 nano-structures in non-stoichiometric grains which grow larger and more numerous with deliberately high electron beam exposures. Such electron beam induced growth of pre-existing nanostructures also appears in the non-stoichiometric areas of devices that have been degraded by the voltage biasing (see ESI Fig. S11†). In addition, after bias degradation we observed a change in grain size and structural re-arrangements for devices fabricated from non-stoichiometric perovskites (5% excess PbI2) as can be seen by comparing the grain structure in Fig. 5c to that in Fig. 5d. | ||
| Fig. 5 Standard cross sectional LVSEM surface morphological images of (a) as-prepared stoichiometric MAPbI3 perovskite; (b) bias degraded stoichiometric MAPbI3 perovskite; (c) as-prepared non-stoichiometric (5 mol% excess PbI2) perovskite; (d) bias degraded non-stoichiometric (5 mol% excess PbI2) perovskite, respectively. | ||
Here, grain boundaries are more numerous and wider after bias degradation (for more information see ESI Fig. S12†). Such structural rearrangement is absent in the devices fabricated from a stoichiometric composition (Fig. 5a and c). The structurally rearranged grains may explain the much reduced short circuit current (Jsc) and PCE for non-stoichiometric devices (ESI Fig. S10 and Table S1†) compared to stoichiometric devices after bias degradation. We also observed other differences in the degradation mechanism(s) between devices made with these different initial compositions which could affect Voc. For the nominally stoichiometric MAPbI3 solar cells, the degradation is initiated near the top electron transport layer (PCBM/MAPbI3 interface) and moves downwards. In contrast, for non-stoichiometric (5% excess PbI2) perovskites, degradation begins near the hole transport layer (NiOx/MAPbI3 interface), see ESI Fig. S13.† The latter might be due to the non-uniform distribution of PbI2 in the PbI2-rich perovskite, which we can clearly see in the as-prepared perovskite materials (bright nano structures near the NiOx/MAPbI3 interface in ESI Fig. S14†). This excess PbI2 at the NiOx/MAPbI3 interface can initiate the decomposition in a similar way to the MAPbI3 pellets with excess PbI2, which degrade faster than the stoichiometric pellets (Fig. 1b and e). Therefore, the presence of excess PbI2 at the NiOx/MAPbI3 interface of PbI2-rich MAPbI3 solar cells might be a reason for such degradation. However, for stoichiometric MAPbI3 we did not find such obvious local excess PbI2 regions prior to degradation. Therefore, in this case we ascribe the degradation resulting from the biasing to the local variations in perovskite composition that might be introduced during the [6,6]-phenyl-C61-butyric acid methyl ester deposition.36 The results above indicate that maintaining a uniform stoichiometry between MAI and PbI2 within individual grains may be an effective approach to enhance the stability of MAPbI3 perovskite materials in the context of charge induced decomposition.
4. Conclusion
Local stoichiometry influences the degradation mechanism of MAPbI3 based perovskite grains. Non-stoichiometric grains degrade faster compared to stoichiometric MAPbI3 under both prolonged air and charge exposure. Furthermore, it can also be seen that devices with excess PbI2 degrade locally when electrically biased in air, which could be explained by a non-uniform distribution of excess PbI2 across the device. Our results suggest that non-uniform stoichiometry is the main cause for localised degradation in perovskite materials. We therefore conclude that any processes which can improve the local and macroscopic homogeneity of MAPbI3 is likely to enhance the overall resistance to degradation in perovskite-structured solar cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for their financial support under projects EP/N008065/1, EP/M025020/1 and EP/L017563/1. V. K. would like to thank W. L. S. (University of Sheffield) and J. B. (Swansea University) for providing perovskite materials and test cells. D. C. S. and I. M. R. thank Innovate UK for funding Knowledge Transfer Project KTP010822. C. L. F. and C. M. H. would like to acknowledge the financial support under EP/P015565/1 and EP/L017563/1 and the use of the University of Sheffield HPC facilities.References
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. Il. Seok, Science, 2017, 356, 1376 CrossRef CAS PubMed.
- Z. K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687 CrossRef CAS PubMed.
- L. Zhang, X. Yang, Q. Jiang, P. Wang, Z. Yin, X. Zhang, H. Tan, Y. Yang, M. Wei, B. R. Sutherland, E. H. Sargent and J. You, Nat. Commun., 2017, 8, 15640 CrossRef CAS PubMed.
- H. Zhu, Y. Fu, F. Meng, X. Wu, Z. Gong, Q. Ding, M. V. Gustafsson, M. T. Trinh, S. Jin and X.-Y. Zhu, Nat. Mater., 2015, 14, 636 CrossRef CAS PubMed.
- S. Shrestha, R. Fischer, G. J. Matt, P. Feldner, T. Michel, A. Osvet, I. Levchuk, B. Merle, S. Golkar, H. Chen, S. F. Tedde, O. Schmidt, R. Hock, M. Rührig, M. Göken, W. Heiss, G. Anton and C. J. Brabec, Nat. Photonics, 2017, 11, 436 CrossRef CAS.
- X. Y. Chin, D. Cortecchia, J. Yin, A. Bruno and C. Soci, Nat. Commun., 2015, 6, 7383 CrossRef CAS PubMed.
- J.-H. Im, I.-H. Jang, N. Pellet, M. Grätzel and N.-G. Park, Nat. Nanotechnol., 2014, 9, 927–932 CrossRef CAS PubMed.
- C. Zuo, H. J. Bolink, H. Han, J. Huang, D. Cahen and L. Ding, Adv. Sci., 2016, 3, 1500324 CrossRef PubMed.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395 CrossRef CAS PubMed.
- S.-G. Li, K.-J. Jiang, M.-J. Su, X.-P. Cui, J.-H. Huang, Q.-Q. Zhang, X.-Q. Zhou, L.-M. Yang and Y.-L. Song, J. Mater. Chem. A, 2015, 3, 9092 RSC.
- Q. Hu, H. Wu, J. Sun, D. Yan, Y. Gao and J. Yang, Nanoscale, 2016, 8, 5350 RSC.
- Z. Yang, C.-C. Chueh, F. Zuo, J. H. Kim, P.-W. Liang and A. K.-Y. Jen, Adv. Energy Mater., 2015, 5, 1500328 CrossRef.
- C. Zuo, D. Vak, D. Angmo, L. Ding and M. Gao, Nano Energy, 2018, 46, 185 CrossRef CAS.
- J. A. Christians, S. N. Habisreutinger, J. J. Berry and J. M. Luther, ACS Energy Lett., 2018, 3, 2136 CrossRef CAS.
- Y. Han, S. Meyer, Y. Dkhissi, K. Weber, J. M. Pringle, U. Bach, L. Spiccia and Y.-B. Cheng, J. Mater. Chem. A, 2015, 3, 8139 RSC.
- Q. Wang, B. Chen, Y. Liu, Y. Deng, Y. Bai, Q. Donga and J. Huang, Energy Environ. Sci., 2017, 10, 516 RSC.
- S.-W. Lee, S. Kim, S. Bae, K. Cho, T. Chung, L. E. Mundt, S. Lee, S. Park, H. Park, M. C. Schubert, S. W. Glunz, Y. Ko, Y. Jun, Y. Kang, H.-S. Lee and D. Kim, Sci. Rep., 2016, 6, 38150 CrossRef CAS PubMed.
- N. Ahn, K. Kwak, M. S. Jang, H. Yoon, B. Y. Lee, J.-K. Lee, P. V. Pikhitsa, J. Byun and M. Choi, Nat. Commun., 2016, 7, 13422 CrossRef CAS PubMed.
- T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubaleb and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 323 RSC.
- Y. Luo, S. Aharon, M. Stuckelberger, E. Magaña, B. Lai, M. I. Bertoni, L. Etgar and D. P. Fenning, Adv. Funct. Mater., 2018, 1706995 CrossRef.
- Q. Ma, S. Huang, S. Chen, M. Zhang, C. F. J. Lau, M. N. Lockrey, H. K. Mulmudi, Y. Shan, J. Yao, J. Zheng, X. Deng, K. Catchpole, M. A. Green and A. W. Y. Ho-Baillie, J. Phys. Chem. C, 2017, 121, 19642 CrossRef CAS.
- Z. Song, S. C. Watthage, A. B. Phillips, B. L. Tompkins, R. J. Ellingson and M. J. Heben, Chem. Mater., 2015, 27, 4612 CrossRef CAS.
- V. Kumar, W. L. Schmidt, G. Schileo, R. C. Masters, W. Wong-stringer, D. C. Sinclair, I. M. Reaney, D. Lidzey and C. Rodenburg, ACS Omega, 2017, 2, 2126 CrossRef CAS.
- D. Prochowicz, P. Yadav, M. Saliba, M. Saski, S. M. Zakeeruddin, J. Lewiński and M. Grätzel, ACS Appl. Mater. Interfaces, 2017, 9, 28418–28425 CrossRef CAS PubMed.
- M. Wong-stringer, O. S. Game, J. A. Smith, T. J. Routledge, B. A. Alqurashy, B. G. Freestone, A. J. Parnell, N. Vaenas, V. Kumar, M. O. A. Alawad, A. Iraqi, C. Rodenburg and D. G. Lidzey, Adv. Energy Mater., 2018, 1801234 CrossRef.
- J. Sun, J. Wu, X. Tong, F. Lin, Y. Wang and Z. M. Wang, Adv. Sci., 2018, 1700780 CrossRef PubMed.
- S. Shrestha, R. Fischer, G. J. Matt, P. Feldner, T. Michel, A. Osvet, I. Levchuk, B. Merle, S. Golkar, H. W. Chen, S. F. Tedde, O. Schmidt, R. Hock, M. Ruhrig, M. Goken, W. Heiss, G. Anton and C. J. Brabec, Nat. Photonics, 2017, 11, 436 CrossRef CAS.
- U. Díaz and A. Corma, Chem.–Eur. J., 2018, 24, 3944 CrossRef PubMed.
- K. J. Abrams, Q. Wan, N. A. Stehling, C. Jiao, A. C. Talari, I. Rehman and C. Rodenburg, Phys. Status Solidi C, 2017, 14, 1700153 Search PubMed.
- J. D. Gale, J. Chem. Soc., Faraday Trans., 1997, 93, 629 RSC.
- C. M. Handley and C. L. Freeman, Phys. Chem. Chem. Phys., 2017, 19, 2313 RSC.
- C. Xiao, Z. Li, H. Guthrey, J. Moseley, Y. Yang, S. Wozny, H. Moutinho, B. To, J. J. Berry, B. Gorman, Y. Yan, K. Zhu and M. Al-Jassim, J. Phys. Chem. C, 2015, 119, 26904 CrossRef CAS.
- S. Wang, Y. Jiang, E. J. Juarez-Perez, L. K. Ono and Y. Qi, Nat. Energy, 2016, 2, 16195 CrossRef.
- H. Yuan, E. Debroye, K. Janssen, H. Naiki, C. Steuwe, G. Lu, M. Moris, E. Orgiu, H. Uji-i, F. D. Schryver, P. Samorì, J. Hofkens and M. Roeffaers, J. Phys. Chem. Lett., 2016, 7, 561 CrossRef CAS PubMed.
- J. Barbé, V. Kumar, M. J. Newman, H. K. H. Lee, S. M. Jain, H. Chen, C. Charbonneau, C. Rodenburg and W. C. Tsoi, Sustainable Energy Fuels, 2018, 2, 905 RSC.
- K. O. Brinkmann, J. Zhao, N. Pourdavoud, T. Becker, T. Hu, S. Olthof, K. Meerholz, L. Hoffmann, T. Gahlmann, R. Heiderhoff, M. F. Oszajca, N. A. Luechinger, D. Rogalla, Y. Chen, B. Cheng and T. Riedl, Nat. Commun., 2017, 8, 13938 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta08231f |
| This journal is © The Royal Society of Chemistry 2018 |