
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic heterojunction photocathodes for optimized photoelectrochemical hydrogen peroxide production†
Maciej
Gryszel
 *ab,
Aleksandr
Markov
ab,
Mikhail
Vagin
ac and
Eric Daniel
Głowacki
ab
*ab,
Aleksandr
Markov
ab,
Mikhail
Vagin
ac and
Eric Daniel
Głowacki
ab
aLaboratory of Organic Electronics, Linköping University, ITN Campus Norrköping, 60221, Norrköping, Sweden. E-mail: maciej.gryszel@liu.se
bWallenberg Centre for Molecular Medicine (WCMM), Linköping University, Linköping, Sweden
cDepartment of Physics, Chemistry and Biology, Linköping University, SE-581 83 Linköping, Sweden
First published on 30th November 2018
Abstract
Solar-to-chemical conversion of sunlight into hydrogen peroxide as a chemical fuel is an emerging carbon-free sustainable energy strategy. The process is based on the reduction of dissolved oxygen to hydrogen peroxide. Only limited amounts of photoelectrode materials have been successfully explored for photoelectrochemical production of hydrogen peroxide. Herein we detail approaches to produce robust organic semiconductor photocathodes for peroxide evolution. They are based on evaporated donor–acceptor heterojunctions between phthalocyanine and tetracarboxylic perylenediimide, respectively. These small molecules form nanocrystalline films with good operational stability and high surface area. We discuss critical parameters which allow fabrication of efficient devices. These photocathodes can support continuous generation of high concentrations of peroxide with faradaic efficiency remaining at around 70%. We find that an advantage of the evaporated heterojunctions is that they can be readily vertically stacked to produce tandem cells which produce higher voltages. This feature is desirable for fabricating two-electrode photoelectrochemical cells. Overall, the photocathodes presented here have the highest performance reported to date in terms of photocurrent for peroxide production. These results offer a viable method for peroxide photosynthesis and provide a roadmap of strategies that can be used to produce photoelectrodes with even higher efficiency and productivity.
Photoelectrochemical conversion of solar energy into chemical fuels is a key emerging sustainable energy approach. Water splitting to generate H2 has been up to now the most popular approach; however, storage and transport of H2 gas present challenges. An alternative concept of storing energy in the form of hydrogen peroxide (H2O2) has been proposed for decades,1,2 yet a few key findings in recent years have allowed this concept to evolve from theory to practice. The first critical finding is the single-compartment peroxide fuel cell, introduced in 2008 and being optimized since.3,4 Peroxide fuel cells have a theoretical efficiency on a par with hydrogen fuel cells. The advantages of H2O2 fuel cells over hydrogen fuel cells are that peroxide is an aqueous solution, promising easier storage and handling, and that they do not require a membrane separator.
The concept of solar energy conversion to hydrogen peroxide in most of the reports is based on photochemical reduction of oxygen to hydrogen peroxide, catalysed by inorganic semiconductors, for example ZnO,5,6 TiO2,7 and CdS.8 In brief, while the oxygenated suspension of an inorganic catalyst is irradiated with an appropriate light source, photogenerated electrons occupying the semiconductor conduction band reduce oxygen, leading to the formation of hydrogen peroxide. The same phenomenon was also shown for materials based on carbon nitride,9,10 and organic semiconductors, recently by our group.11 As the reductive part of the photochemical process is efficient enough to lead to the accumulation of hydrogen peroxide up to a few mM, the major challenge is its oxidative part, necessary for closing the redox cycle. In most cases the energy level of the semiconductor valence band lies too high to enable water oxidation by the photoinduced hole. It can not only limit the system performance towards H2O2 evolution but also significantly affects the catalyst stability, as it can undergo self-oxidation.11 Although the possibility of a full redox cycle leading to photoinduced oxygen reduction to hydrogen peroxide with simultaneous water oxidation was reported for some of the wide-bandgap semiconductors,12,13 the most common practice is the addition of sacrificial electron donors (e.g. oxalate and formate) to the reaction system. The oxidation potentials of these compounds are much lower than that of water, which allows the process to occur even for lower-bandgap semiconductors, such as polythiophene. Another possibility of overcoming the water oxidation limitation is photoelectrocatalysis. The field, first explored in 1972 by Fujishima and Honda14 using a TiO2 based photoanode for water oxidation with hydrogen evolution on a platinum counter electrode, has developed significantly since that time. In comparison to photocatalysis, a photoelectrochemical cell allows one to separate reduction and oxidation processes to two different materials, each optimized for the intended electrochemical transformation. The possible application of a voltage bias can also facilitate the process and overcome the thermodynamic and kinetic barriers related to water oxidation. Although many examples of photocathodes15–17 and photoanodes18,19 enabling water splitting to H2 and O2 were published, the concept of a photocathode for hydrogen peroxide remains relatively unexplored. In the first report of such a device, Jakešová et al. showed that an organic semiconductor, the yellow pigment epindolidione (EPI), deposited on a gold substrate, is able to promote H2O2 photosynthesis, giving a stable photocurrent of approx. 100 μA cm−2 over the course of 48 h with 96% faradaic efficiency.20 This work was followed by a similar report on devices based on biscoumarin-containing acenes.21 Recently, photocathodes based on porphyrins,22,23 chemically polymerized eumelanin,24 polymeric metal salen-type complexes,25 and dye-sensitized NiO were reported.26 Despite high faradaic yields achieved by these devices, a key parameter, photocurrent density, should be improved in order to achieve a H2O2 evolution rate sufficient enough for effective solar energy harvesting. While top-performing photocathodes for hydrogen evolution, biased at 0 V vs. Ag/AgCl, achieve photocurrent densities of up to 22 mA cm−2 under 1 sun irradiation,27 the abovementioned photocathodes for H2O2 photosynthesis give a much lower photocurrent density (around 0.1 mA cm−2) under these conditions; therefore there is a possibility of progress in this field. The realistic upper limit for the photocurrent in such a cell is limited by the concentration and diffusion of dissolved O2; however values of several mA cm−2 should be possible.
Herein we present our work on photocathodes for H2O2 synthesis based on an organic donor–acceptor heterojunction (referred to hereafter as the PN junction) made by evaporation of metal-free phthalocyanine (H2Pc) and N,N′-dimethyl perylenetetracarboxylic bisimide (PTCDI), respectively (Fig. 1). This is the first example of H2O2 evolving photoelectrodes based on an organic PN junction, despite wide utilization of this concept in photovoltaics and photoelectrocatalysis for hydrogen evolution.28,29 The concept of the H2Pc/PTCDI photocathode was based on previous findings by our group. Organic PN junctions made from these materials were recently reported as showing the ability to be charged upon excitation with pulsed red light (≤5 ms) in an electrolytic environment, producing stable devices for neuronal stimulation.30 Although this process was proved to be non-faradaic in nature under these conditions, the PN junction behaviour under constant irradiation with white light was not investigated. In a parallel study, we also proved that if PTCDI films are polarized cathodically in an oxygenated electrolyte, they are able to electrochemically produce H2O2, being completely stable despite hundreds of hours of operation at high current density.31 As the PTCDI on Au system cannot be used as a photocathode due to the very low p-type conductivity of this semiconductor, the H2Pc/PTCDI heterojunction, ambipolar as a whole, gives the opportunity to obtain the photocathode which utilises favourable catalytic properties of the PTCDI towards the O2 to H2O2 reduction.
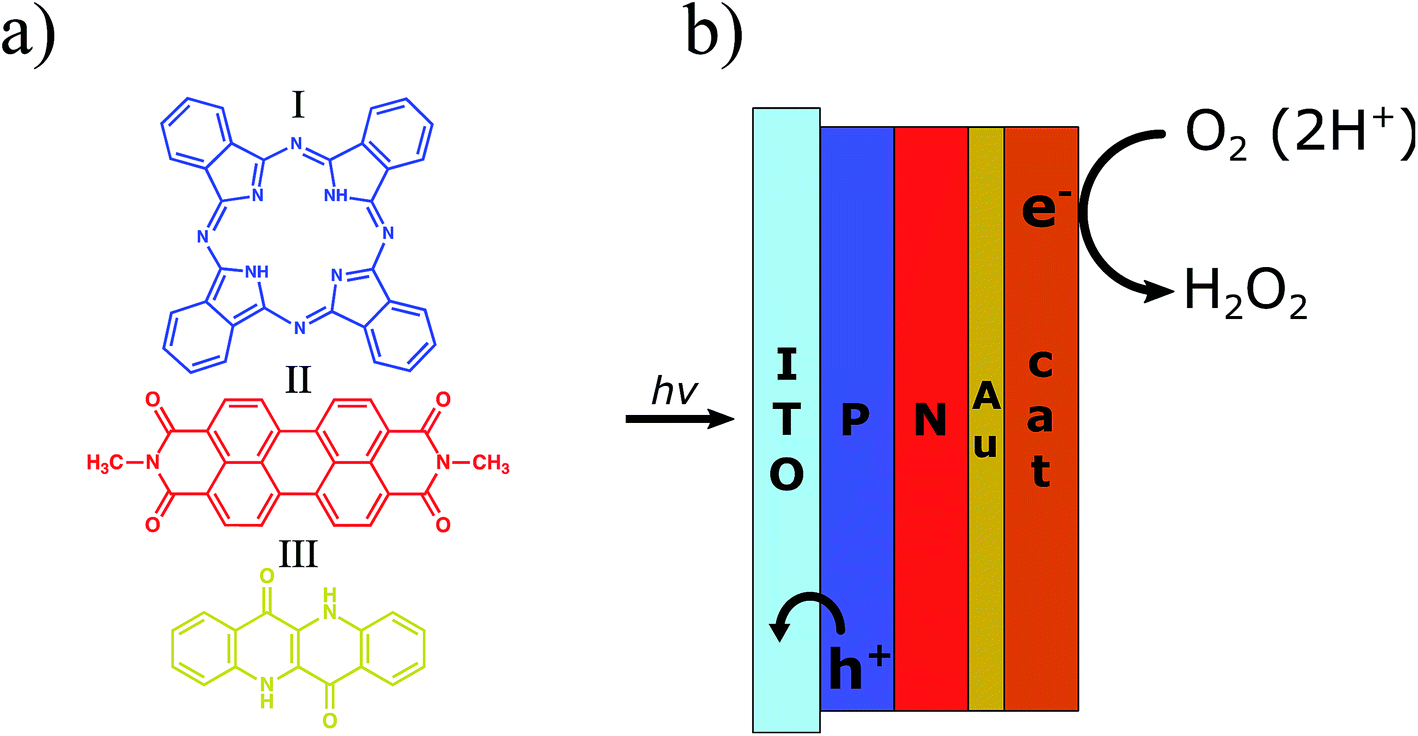 | ||
| Fig. 1 (a) Chemical structure of the organic semiconducting pigments used in this work for fabrication of photocathodes: I – metal-free phthalocyanine (H2Pc), II – N,N′-dimethyl perylenetetracarboxylic diimide (PTCDI), and III – epindolidione (EPI). The outline colour corresponds to the approximate colour of the pigment. (b) Schematic of the PN/Au/catalyst photocathodes. H2Pc (P), PTCDI (N) and Au are used as the organic donor, the organic acceptor, and electron transporting layer, respectively. The catalyst layer is either PTCDI or EPI. The thicknesses of the layers in the best performing devices characterized herein were as follows: 60/60/5/30 nm for the PN/Au/PTCDI photocathode and 30/30/5/100 nm for the PN/Au/EPI photocathode. | ||
We first evaluated the photoelectrochemical properties of the H2Pc/PTCDI junction. This we fabricated by subsequent vacuum evaporation of both pigments on indium tin oxide (ITO) substrates modified with n-octyltriethoxysilane (OTS), resulting in the 60/60 nm double layer organic heterojunction with a nanocrystalline morphology. For comparison, H2Pc on ITO and EPI on ITO were investigated as well. These samples were electrochemically characterized by cyclic voltammetry and chronoamperometry in a three-electrode system with a Ag/AgCl reference electrode (Fig. 2a and b), using an electrochemical H-cell as shown in Fig. S1.† The measurements were performed either under a tungsten halogen lamp illumination of 100 mW cm−2 intensity or without illumination. All details of the sample preparation and electrochemical characterization can be found in the ESI.† As expected, the H2Pc/PTCDI photocathode (hereinafter PN photocathode), biased at 0 V vs. Ag/AgCl under chopped illumination with 15 s amplitude, showed a pulsed photocurrent response of a constant value, proving the existence of a photofaradaic process. The system performance turned out to be similar to that of a reference EPI photocathode, prepared and measured in the same way. As the optical characteristics of the PN allow for far superior light absorption compared with EPI and it is known that PTCDI is a good catalyst for the O2 to H2O2 reduction, the limited performance of the PN photocathode suggests that charge carrier recombination limits the performance. To suppress this process, we decided to deposit a thin layer of Au as an electron accepting layer on top of the PN photocathode. Besides being a good ohmic contact for electrons of PTCDI, Au at the same time can be a good catalyst for O2/H2O2 reduction.32–34 The PN/Au photocathode showed an impressive, roughly twenty-fold photocurrent increase, to over 800 μA cm−2 at 0 V vs. Ag/AgCl bias, stable for at least 2 h (black traces, Fig. 2a and b). However, under these conditions (pH 2) the faradaic yield was low (32.5–18.3%), regardless of the Au layer thickness used. Apparently, the gold structure obtained by vacuum evaporation does not favour the selective oxygen reduction to hydrogen peroxide under these conditions. The faradaic yield is lowered by the 4e− reduction of O2 to H2O, 2e− reduction of H2O2 to H2O, or competition from H2 evolution. In the next step of the photocathode optimization, we deposited a more selective electrocatalyst for H2O2 electrosynthesis on the PN/Au structure. There are many examples of such catalysts in the literature;35–37 nevertheless, based on our previous studies, we chose to fabricate two different PN photocathodes with organic pigments as catalysts for H2O2 electrosynthesis: PTCDI and EPI. The structures of the final photocathodes discussed in this work, PN/Au/PTCDI and PN/Au/EPI, are illustrated in Fig. 1b. It is worth noting that in the case of the PN/Au/PTCDI photocathode there are two layers of PTCDI which play different roles. The first one, deposited directly on H2Pc, works as an n-type semiconductor in the organic PN junction. The second, final PTCDI layer is the electrocatalyst for the H2O2 synthesis. Compared with the PN/Au photocathode, the PN/Au/PTCDI, and PN/Au/EPI photocathodes showed significantly improved faradaic yields without almost any drop in photocurrent, giving stable photoelectrodes having the characteristic shown in Fig. 2c and d. The cyclic voltammetry experiments proved that both systems demonstrate high activity towards photoelectrochemical reduction of oxygen within a wide pH (pH = 2, 7, and 12) and bias potential range. For both photocathodes, the highest photocurrent was registered for the acidic electrolyte, what is consistent with previous findings for the PTCDI on Au electrode31 and EPI on Au photoelectrode.20 Chronoamperometry experiments at 0 V vs. Ag/AgCl bias with 15 s light on/off cycles show excellent carrier transport dynamics, with a constant current value within the whole 15 s cycle. Lack of transient peaks on the I = f(t) plot proves that these systems do not suffer from the occurrence of trap states (Fig. 2b, green and red traces).
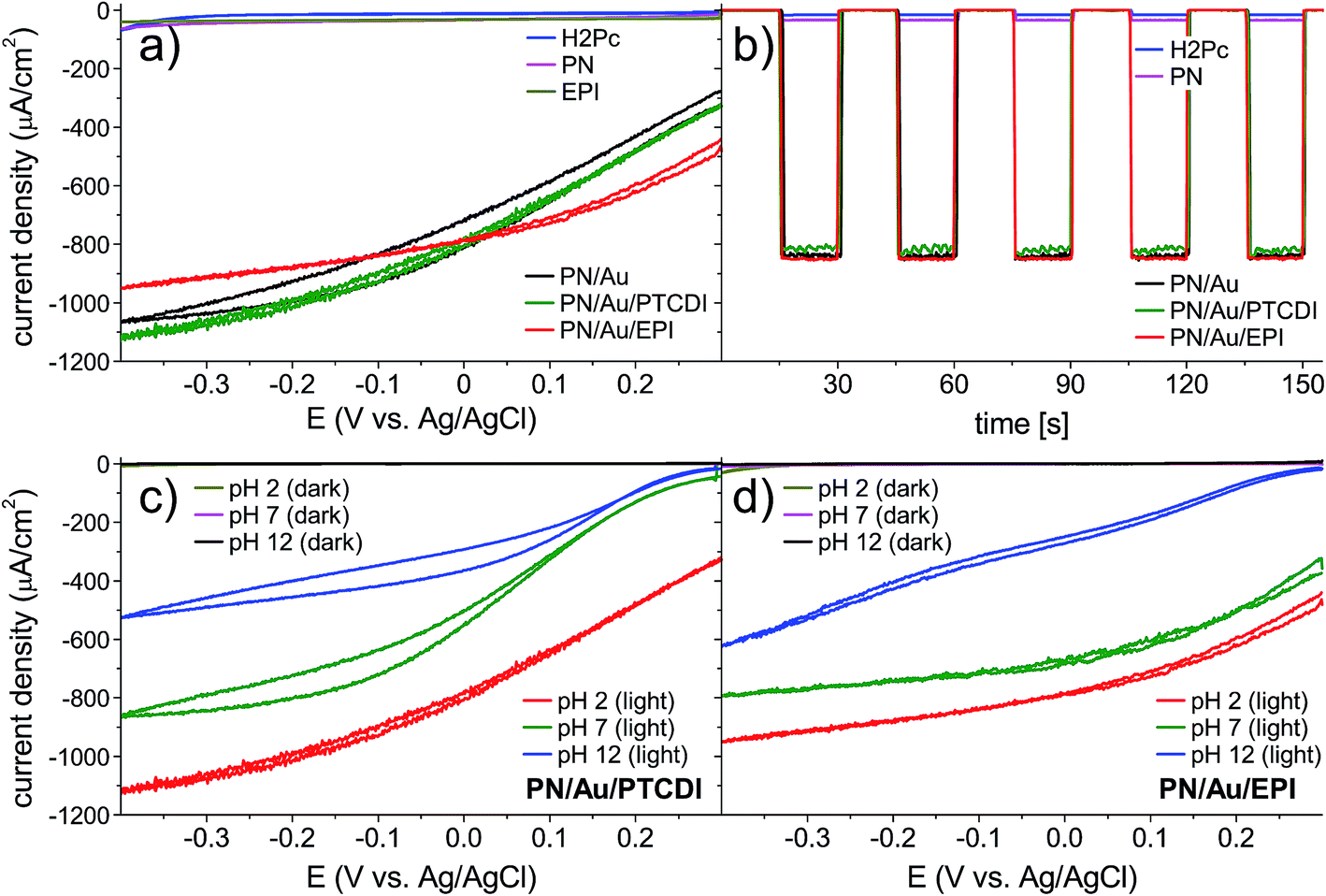 | ||
| Fig. 2 (a) Cyclic voltammetry of the photocathodes at pH 2 under 100 mW cm−2 irradiation. As a reference, EPI and H2Pc covered ITO substrates are prepared and measured. (b) Chronoamperometry of the photocathodes at pH 2 and 0 V vs. Ag/AgCl under pulsed (15 s) 100 mW cm−2 irradiation. (c and d) Comparison of cyclic voltammograms for the PN/Au/PTCDI and PN/Au/EPI photocathodes at pH 2, pH 7, and pH 12 in the dark and under 100 mW cm−2 irradiation. | ||
To get a more detailed picture of the performance of the photoelectrochemical system, we performed linear sweep voltammetry with 15 s light on/off cycles in the −0.4 V to +0.8 V vs. Ag/AgCl range for PN/Au/PTCDI (Fig. 3a) and PN/Au/EPI (Fig. 3b). With these measurements we can estimate the onset potential of the photoelectrochemical process, and thus accurately determine the photovoltage generated by the device. The calculated onset potentials are shown in the insets in Fig. 3a and b. As expected, in both cases the highest onset potential (defined as the potential at which the photocurrent density is at least 10 μA cm−2) values were obtained at pH 2, being equal to 0.61 V and 0.68 V vs. Ag/AgCl for the PN/Au/PTCDI and PN/Au/EPI, respectively. As the obtained values are close to the theoretical 0.74 V vs. Ag/AgCl (assuming that the maximum photovoltage generated by the H2Pc/PTCDI PN junction is 550 mV,38 the oxidation potential of the 2H+ + 2e− + O2 ⇌ H2O2 process is 0.19 V at pH 2 vs. Ag/AgCl4 and 0 V overpotential of oxygen reduction), it proves not only a judicious selection of the oxygen reduction catalysts used, but also the 2e−/2H+ O2 reduction mechanism itself. In the case of photoelectrochemical formation of hydrogen peroxide in acidic solution, there are two possible pathways of oxygen reduction leading to peroxide: direct 2e−/2H+ reduction (2H+ + 2e− + O2 ⇌ H2O2), or 1e− reduction of oxygen to a protonated superoxide radical, which subsequently disproportionates to H2O2 (H+ + e− + O2 ⇌ HO2˙ and then 2HO2˙ ⇌ H2O2 + O2). Two-electron reduction, while thermodynamically favourable, may be kinetically more demanding than the single-electron pathway. As the oxidation potential of the protonated superoxide radical at pH 2 is −0.215 V vs. Ag/AgCl,39 a photocathode for 1e− reduction of oxygen to superoxide would not generate any photocurrent when the bias potential is more positive than 0.4 vs. Ag/AgCl. Based on the obtained onset potential values, corroborated by the evidence of the faradaic yield measurements showing efficient H2O2 generation, we can conclude that the 2e−/2H+ reduction mechanism of O2 to H2O2 is the dominant pathway.
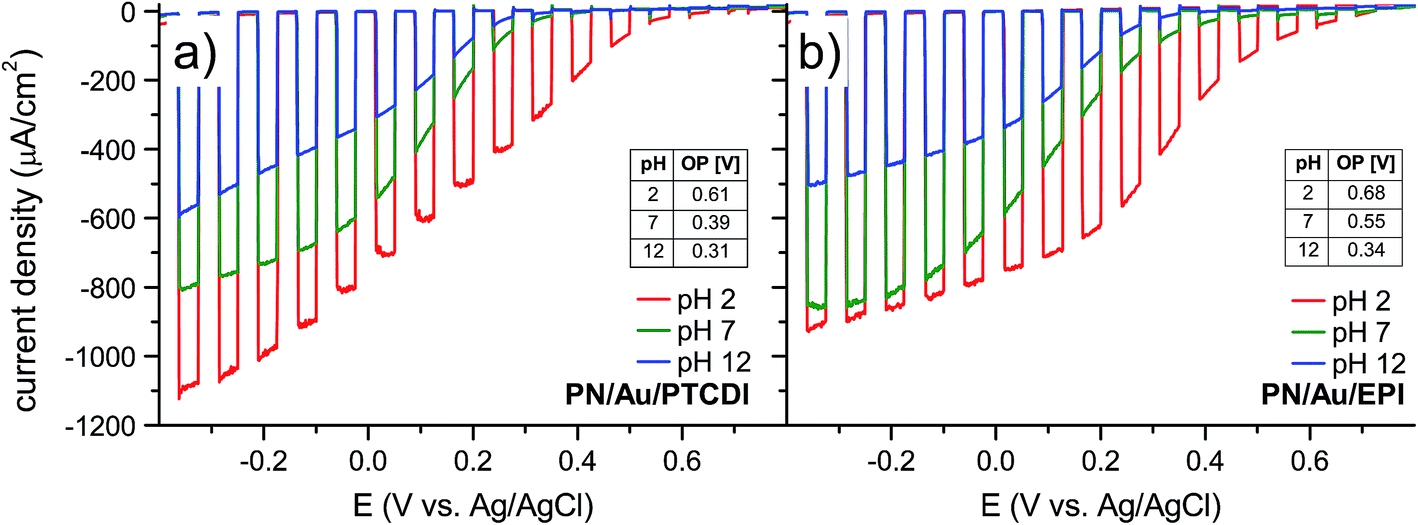 | ||
| Fig. 3 Linear sweep voltammetry with 15 s light on/off cycles for (a) PN/Au/PTCDI and (b) PN/Au/EPI. Scans were measured at pH 2, pH 7, and pH 12 with 100 mW cm−2 irradiation. Values of onset potential (OP) at a given pH are shown in the inset tables. | ||
To provide additional evidence that our deduction of the 2e−/2H+ mechanism is correct, we performed Koutecký–Levich analysis with a rotating disc electrode (RDE) allowing the determination of the number of electrons transferred per oxygen molecule. To avoid any possible issues with photocurrent stability (a reliable calculation requires very stable current), we simplified the system by measuring the electrocatalytic layer of 30 nm of PTCDI alone, which we deposited on the glassy carbon RDE surface. The high stability of the PTCDI performance for the O2 to H2O2 reduction was proven before.31 We conducted a series of linear sweep voltammetry scans with different rotation speeds in the oxygenated pH 2 electrolyte in the +0.4 V to −0.5 V vs. Ag/AgCl range (Fig. S2†). The details of the RDE experiment and Koutecký–Levich analysis can be found in the ESI.† The number of electrons transferred per oxygen molecule was calculated to be 2.03 at the −0.5 V vs. Ag/AgCl bias, which confirms the conclusion of the 2e−/2H+ pathway considering the onset potential values.
Next, longer term photoelectrolysis experiments (6+ hours) were conducted with PN/Au/PTCDI and PN/Au/EPI systems and the faradaic efficiency of H2O2 production was checked. During these experiments, the electrolyte was constantly purged with moist O2 gas and the catholyte was stirred. We found this procedure to be critical for maintaining a high oxygen reduction photocurrent (Fig. S3†). Both photoelectrodes, biased at 0 V vs. Ag/AgCl, showed a slight photocurrent decrease over time, and yet still retained 75% of their initial photocurrent after 6 h of photoelectrolysis at pH 2 (Fig. 4a and b). Within 6 h of photoelectrolysis, both PN/Au/PTCDI and PN/Au/EPI photocathodes showed good faradaic yield (86–62%, as shown in Fig. 4a and b); however, the values decreased over the course of the experiments. Previously, we observed the same dependence for the PTCDI on Au cathodes and it is related to the fact that a long electrolysis process results in the accumulation of H2O2, whose concentration is comparable to the concentration of O2 (approx. 1.3 mM under 1 atm). Under these conditions, H2O2 reduction to H2O competes with the process of O2/H2O2 synthesis and thus causes a decrease in the faradaic yield. For the same PTCDI on Au cathode it was also observed that the lower the current density, the higher the faradaic yield. This can be a good explanation for the lower faradaic yield of the PN/Au/EPI system in comparison to the previously reported EPI photocathodes. In that case, the faradaic yield was 96%; however, the current density was over 8 times lower than that for the PN/Au/EPI photocathode tested in this work.
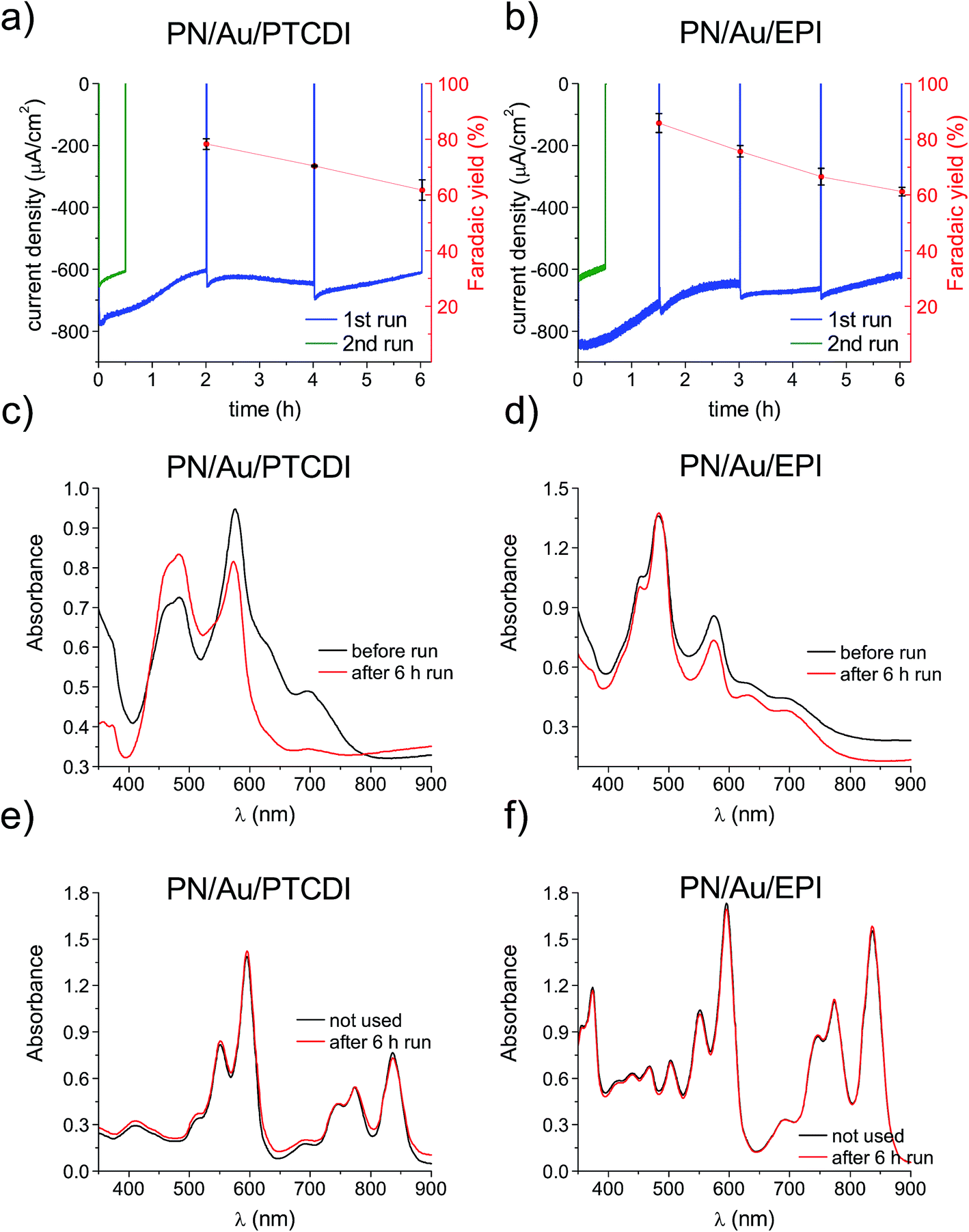 | ||
| Fig. 4 Photoelectrolytic H2O2 evolution experiments for (a) PN/Au/PTCDI and (b) PN/Au/EPI photocathodes at pH 2, 0 V vs. Ag/AgCl bias, and under 100 mW cm−2 irradiation. The blue traces show the first 6 h run for the given photocathode while the green ones correspond to the second run. (c and d) Comparison of the UV-vis spectra for the respective photocathodes before and after 6 h of photoelectrolysis. (e and f) Comparison of the UV-vis spectra for the organic components of the same photocathodes dissolved in conc. H2SO4 after 6 h of photoelectrolysis with the corresponding spectra obtained for fresh devices. While optical absorption changes are obvious in the thin films, any optical changes are absent when the films are dissolved. This suggests that a morphological degradation and not a chemical degradation occurs. | ||
Repetition of the experiment with the already 6 hour used samples showed that the performance loss is not reversible (Fig. 4a and b; green traces). As any physical damage to the layers (e.g. delamination or swelling) was not readily apparent, the performance loss could not be explained by the shrinking of the active area. Degradation of ITO used as a conducting substrate can also be excluded, as the same performance decrease was also observed when FTO and thin, transparent Au substrates were used. We compared SEM images of the samples before and after the long photoelectrolysis experiment to see if the observed phenomenon can be explained by the change in the catalyst layer morphology (Fig. S4†). However, in neither case had the morphology (shape and size of the nanocrystallites) changed obviously. That led to the conclusion that the drop in the photocathode performance may have been caused by a change in the crystalline structure of the organic pigment layers or irreversible chemical degradation of the multi-layered photoelectrocatalytic system. Organic pigments are widely known for their occurrence in different crystalline forms, differing in their optical properties.40 Polymorphic transition can be induced by many factors, such as solvent treatment,41 post deposition annealing42 or light exposure.43 This can result in changes in optical properties and charge carrier mobilities and is likely in the case of our devices, operating under water conditions under constant illumination. Therefore, UV-vis spectra of the photocathodes before and after 6 h of photoelectrolysis were measured and compared (Fig. 4c and d). A significant change could be seen, especially for H2Pc related peaks. This could be the result of the polymorphic transition, but could also be explained by chemical degradation. The latter seemed to be less likely, as in the case of chemical degradation of the layer deposited directly on the conducting substrate, its chemical degradation would probably mean delamination of the whole multilayered system. The possibility of chemical degradation was excluded by measuring the optical absorption of the pigments following dissolution of the device layers in conc. H2SO4. Many insoluble organic pigments can be dissolved in H2SO4, as the strong acid protonates carbonyl moieties.44 In the discussed case it can give a true picture of the chemical composition of the photocathodes without the influence of the polymorphic transitions. The UV-vis spectra of the H2SO4 dissolved cathodes show no change compared with the spectra of the corresponding solutions obtained for the unused samples. This proves that the photocathodes presented in this work are not affected by chemical degradation within 6 h of photoelectrolysis. This is an important finding in light of our previous studies,11 confirming the hypothesis that the very same organic semiconductor, which is unstable as a photocatalyst even in the presence of a sacrificial electron donor, can be significantly more chemically stable after its deposition on the conductive substrate, when the applied bias facilitates efficient extraction of holes.
The UV-vis measurements of the photocathodes before and after the 6 h of photoelectrolysis demonstrated that the most probable explanation of the performance drop of the photocathodes is polymorphic transition of their constituents. However, the possibility of the problem with metastable PN interface morphology cannot be excluded. This is a common issue in organic bulk heterojunction solar cells.45 As described by Schaffer et al. with the example of a P3HT:PCBM based solar cell, the morphological degradation and performance worsening of the device during its operation is caused by the phase separation and growth of the domain size of the polymeric donor.46 Although the photocathodes discussed herein, prepared by the vacuum evaporation process, are not typical bulk heterojunction structures, the interface morphology of the devices can also be crucial for their efficient performance. A thorough explanation of the changes leading to instability in these photocathode devices, supported by the structural investigation, will be provided in a forthcoming study.
The satisfactory performance of the devices operating at 0 V vs. Ag/AgCl at pH 2 encouraged us to check the possibility of their application in a nonbiased, 2-electrode system with a Pt counter electrode (Fig. 5). Unfortunately, upon irradiation, the photocurrent, low from the beginning, quickly vanished. We hypothesized that the process is limited by the relatively high potential of water oxidation at the Pt counter electrode and that the issue can be solved by changing the pH of the solution, as the reaction potential is pH dependent (i.e. 1.23 V at pH 0, 0.817 V at pH 7). To determine the optimum pH value for the 2-electrode photoelectrochemical cell, we ran the chronoamperometry experiment with pulsed light and a −0.5 V vs. Pt counter electrode bias, which was enough to register the photocurrent even for the pH 2 solution. As shown in Fig. 4b, in contrast to the previously determined photocurrent dependence for the photocathodes operating at 0 V vs. Ag/AgCl in electrolytes of different pH, for the 2-electrode setup, as expected, at the same bias, the higher the pH the higher the photocurrent. Therefore, the photoelectrochemical cell operation is limited by water oxidation, and not the photocathodic process. Despite switching to the basic electrolyte, the photocurrent under non-biased conditions was still below 100 nA. It was clear that the photovoltage generated by the photoelectrode was too low. To address this issue, we fabricated the PN/Au/PTCDI tandem photocathodes with multiple PN junctions (Fig. 5a). The concept was inspired by previously published studies on so-called artificial leaves for photochemical hydrogen evolution – photovoltaic devices with multiple semiconductor layers which are able to generate a photovoltage high enough to run the water splitting reaction.47,48 We deposited the Au/PTCDI layers on top of the PN junction stacks with different number of junctions. As a recombination contact between the PN junctions, a transparent, thin Au layer (2 nm) was used in all cases. The performance of the devices with 1, 2, and 3 PN layers, operating in a 2 electrode setup at a −0.3 V bias vs. a Pt counter electrode, is shown in Fig. 5c. The 2PN/Au/PTCDI device gives an almost 3-fold photocurrent increase compared with the initial PN/Au/PTCDI design. The additional, third PN junction, gives a further rise in the performance and enables the possibility of running the photoelectrolysis process in a two-electrode setup without any external bias (Fig. 5d). Nevertheless, the device performance still needs to be improved, as the photocurrent is relatively low and unstable (over 50% drop within 3 h). However, the concept of tandems is viable, and is easily applicable in the case of evaporated thin films.
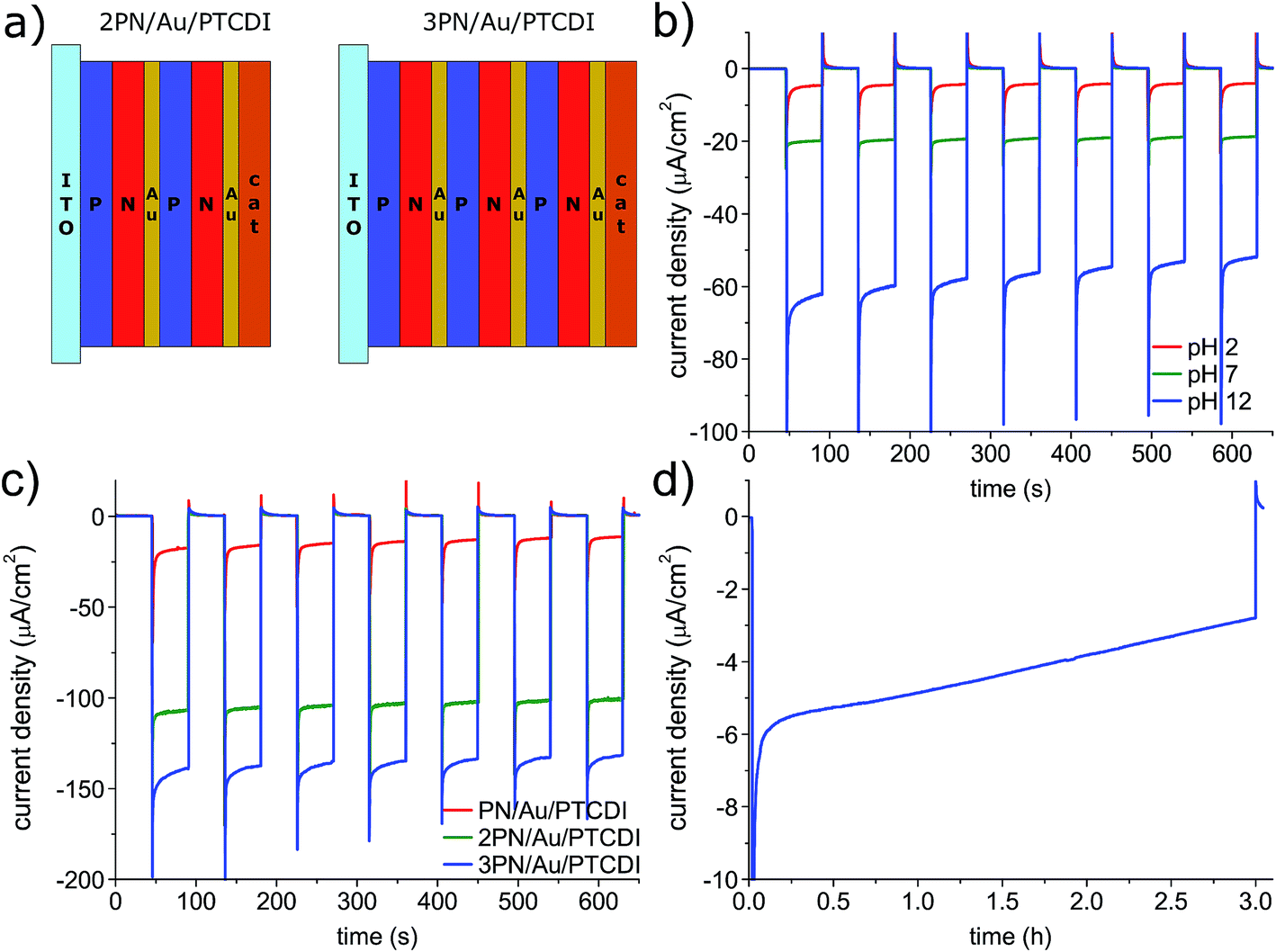 | ||
| Fig. 5 (a) Structure of the tandem photocathodes. The optimum PN thickness was determined to be 30/30 nm; thicker layers prevent efficient light capture in the terminal PN junction in the 3 PN device, which worsens the performance. (b) The pH dependence for the PN/Au/PTCDI device operating in a 2-electrode arrangement at a constant bias of −0.5 V vs. a Pt counter electrode under pulsed (45 s) 100 mW cm−2 irradiation. In all cases, electrolyte with the same ionic strength was used (0.1 M). (c) Comparison of the tandem devices with different number of PN junctions operating in a 2-electrode arrangement at a constant bias of −0.5 V vs. a Pt counter electrode under pulsed (45 s) 100 mW cm−2 irradiation. The PN/Au/PTCDI sample used in this experiment has 30/30 nm PN junction thickness and was prepared only as a reference for the 2 and 3 PN devices. (d) The I = f(t) dependence for a longer photoelectrolysis experiment with the 3 PN photocathode without external bias at pH 12. | ||
In summary, photocathodes with an organic semiconductor heterojunction and catalysts based on organic pigments were demonstrated as efficient devices for H2O2 photosynthesis. The photocurrent generated by the PN/Au/PTCDI and PN/Au/EPI devices at 0 V vs. Ag/AgCl is the highest ever reported for this process. The materials comprising the devices are chemically stable and do not apparently undergo any chemical degradation; however, their long-term performance is affected by morphological degradation. Though a long-standing belief that organic semiconductors are inherently photochemically unstable under oxygenated and aqueous conditions still exists, recent research has suggested that this is not necessarily the case. Crystalline pigments, such as those used here, with stable heteroatomic substituents and aromatic structures are known from the field of industrial colorants to show outstanding stability. Here we see that the organic materials do not chemically degrade, but rearrange morphologically, resulting in a decline in performance. Morphological changes such as donor/acceptor demixing have been implicated as the primary problem in organic heterojunction photovoltaics. It is therefore likely that the same issue needs to be addressed in organic photoelectrodes. This work proves that the discussed structures may be easily fabricated from cheap and abundant materials (such as the industrial pigments used in this work); however, further progress, addressing the issue of long-term stability and maximising the faradaic yield of the process, still needs to be made. Another aspect of morphological optimisation is the utilization of a bulk heterojunction architecture created by coevaporation of donor and acceptor materials, or potentially finding solution-processable analogues which also allow such a heterojunction formation. Application of other semiconductors and reaction catalysts should be evaluated, as it could not only increase the long-term stability and the reaction selectivity, but also the device photovoltage, enabling possibility of efficient photoelectrolysis without external bias.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors are grateful for financial support from the Knut and Alice Wallenberg Foundation, especially from the Wallenberg Centre for Molecular Medicine at Linköping University, and Vinnova within the framework of Treesearch.se.References
- R. S. Disselkamp, Energy Fuels, 2008, 22, 2771–2774 CrossRef CAS.
- R. S. Disselkamp, Int. J. Hydrogen Energy, 2010, 35, 1049–1053 CrossRef CAS.
- S. ichi Yamazaki, Z. Siroma, H. Senoh, T. Ioroi, N. Fujiwara and K. Yasuda, J. Power Sources, 2008, 178, 20–25 CrossRef.
- S. Fukuzumi and Y. Yamada, ChemElectroChem, 2016, 3, 1978–1989 CrossRef CAS.
- T. R. Rubin, J. G. Calvert, G. T. Rankin and W. MacNevin, J. Am. Chem. Soc., 1953, 75, 2850–2853 CrossRef CAS.
- A. J. Hoffman, E. R. Carraway and M. R. Hoffmann, Environ. Sci. Technol., 1994, 28, 776–785 CrossRef CAS PubMed.
- J. R. Harbour, J. Tromp and M. L. Hair, Can. J. Chem., 1985, 63, 204–208 CrossRef CAS.
- R. E. Stephens, B. Ke and D. Trivich, J. Phys. Chem., 1955, 59, 966–969 CrossRef CAS.
- Y. Kofuji, Y. Isobe, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, J. Am. Chem. Soc., 2016, 138, 10019–10025 CrossRef CAS PubMed.
- R. Wang, X. Zhang, F. Li, D. Cao, M. Pu, D. Han, J. Yang and X. Xiang, J. Energy Chem., 2018, 27, 343–350 CrossRef.
- M. Gryszel, M. Sytnyk, M. Jakešová, G. Romanazzi, R. Gabrielsson, W. Heiss and E. D. Głowacki, ACS Appl. Mater. Interfaces, 2018, 10, 13253–13257 CrossRef CAS PubMed.
- N. Kaynan, B. A. Berke, O. Hazut and R. Yerushalmi, J. Mater. Chem. A, 2014, 2, 13822–13826 RSC.
- T. Baran, S. Wojtyła, A. Vertova, A. Minguzzi and S. Rondinini, J. Electroanal. Chem., 2018, 808, 395–402 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed.
- L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48, 988–990 RSC.
- J. D. Benck, S. C. Lee, K. D. Fong, J. Kibsgaard, R. Sinclair and T. F. Jaramillo, Adv. Energy Mater., 2014, 4(18), 1400739 CrossRef.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- M. Jakešová, D. H. Apaydin, M. Sytnyk, K. Oppelt, W. Heiss, N. S. Sariciftci and E. D. Głowacki, Adv. Funct. Mater., 2016, 26, 5248–5254 CrossRef.
- M. K. Węcławski, M. Jakešová, M. Charyton, N. Demitri, B. Koszarna, K. Oppelt, S. Sariciftci, D. T. Gryko and E. D. Głowacki, J. Mater. Chem. A, 2017, 20780–20788 RSC.
- N. U. Day and C. C. Wamser, J. Phys. Chem. C, 2017, 121, 11076–11082 CrossRef CAS.
- D. H. Apaydin, H. Seelajaroen, O. Pengsakul, P. Thamyongkit, N. S. Sariciftci, J. Kunze-Liebhuser and E. Portenkirchner, ChemCatChem, 2018, 10330, 1793–1797 CrossRef PubMed.
- L. Migliaccio, M. Gryszel, V. Đerek, A. Pezzella and E. D. Głowacki, Mater. Horiz., 2018, 5, 984–990 RSC.
- A. S. Konev, Y. Kayumov, M. P. Karushev and Y. V. Novoselova, ChemElectroChem, 2018, 5, 1–6 Search PubMed.
- O. Jung, M. L. Pegis, Z. Wang, G. Banerjee, C. T. Nemes, W. L. Hoffeditz, J. T. Hupp, C. A. Schmuttenmaer, G. W. Brudvig and J. M. Mayer, J. Am. Chem. Soc., 2018, 140, 4079–4084 CrossRef CAS PubMed.
- B. Seger, T. Pedersen, A. B. Laursen, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, J. Am. Chem. Soc., 2013, 135, 1057–1064 CrossRef CAS PubMed.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, S. Campidelli, R. Cornut and B. Jousselme, J. Mater. Chem. A, 2016, 4, 4831–4839 RSC.
- A. Ghadirzadeh, F. Fumagalli, A. Mezzetti, S. Bellani, L. Meda, M. R. Antognazza and F. Di Fonzo, ChemPhotoChem, 2018, 2, 283–292 CrossRef CAS.
- D. Rand, M. Jakešová, G. Lubin, I. Vebraite, M. David-Pur, V. Derek, T. Cramer, N. S. Sariciftci, Y. Hanein and E. D. Głowacki, Adv. Mater., 2018, 30, 1707292 CrossRef PubMed.
- M. Warczak, M. Gryszel, M. Jakešová, V. Derek and E. D. Głowacki, Chem. Commun., 2018, 54, 1960–1963 RSC.
- Y. Yang, C. Dai, A. Fisher, Y. Shen and D. Cheng, J. Phys.: Condens. Matter, 2017, 29, 365201 CrossRef PubMed.
- N. S. K. Gowthaman, S. Shankar and S. Abraham John, J. Electroanal. Chem., 2018, 812, 37–44 CrossRef CAS.
- E. Pizzutilo, O. Kasian, C. H. Choi, S. Cherevko, G. J. Hutchings, K. J. J. Mayrhofer and S. J. Freakley, Chem. Phys. Lett., 2017, 683, 436–442 CrossRef CAS.
- Y. Sun, I. Sinev, W. Ju, A. Bergmann, S. Dresp, S. Kühl, C. Spöri, H. Schmies, H. Wang, D. Bernsmeier, B. Paul, R. Schmack, R. Kraehnert, B. Roldan Cuenya and P. Strasser, ACS Catal., 2018, 8, 2844–2856 CrossRef CAS.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef.
- H. W. Kim, M. B. Ross, N. Kornienko, L. Zhang, J. Guo, P. Yang and B. D. McCloskey, Nat. Catal., 2018, 1, 282–290 CrossRef.
- P. Peumans, A. Yakimov and S. R. Forrest, J. Appl. Phys., 2003, 93(7), 3693–3723 CrossRef CAS.
- D. A. Armstrong, R. E. Huie, S. Lymar, W. H. Koppenol, G. Merényi, P. Neta, D. M. Stanbury, S. Steenken and P. Wardman, BioInorg. React. Mech., 2013, 9, 59–61 CAS.
- G. Klebe, F. Graser, E. Hädicke and J. Berndt, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 69–77 CrossRef.
- Q. Zhang, H. Chen, Y. Liu and D. Huang, Dyes Pigm., 2004, 63, 11–16 CrossRef CAS.
- S. M. Bayliss, S. Heutz, G. Rumbles and T. S. Jones, Phys. Chem. Chem. Phys., 1999, 1, 3673–3676 RSC.
- L. Gaffo, M. J. S. P. Brasil, F. Cerdeira and W. C. Moreira, Thin Solid Films, 2005, 488, 236–241 CrossRef CAS.
- Y. Nagao, Prog. Org. Coat., 1997, 31, 43–49 CrossRef CAS.
- P. Cheng and X. Zhan, Chem. Soc. Rev., 2016, 45, 2544–2582 RSC.
- C. J. Schaffer, C. M. Palumbiny, M. A. Niedermeier, C. Jendrzejewski, G. Santoro, S. V. Roth and P. Müller-Buschbaum, Adv. Mater., 2013, 25, 6760–6764 CrossRef CAS PubMed.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed.
- S. Esiner, R. E. M. Willems, A. Furlan, W. Li, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2015, 3, 23936–23945 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta08151d |
| This journal is © The Royal Society of Chemistry 2018 |