
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
IrOOH nanosheets as acid stable electrocatalysts for the oxygen evolution reaction†
Daniel
Weber‡
 ab,
Leslie M.
Schoop
c,
Daniel
Wurmbrand
d,
Sourav
Laha
a,
Filip
Podjaski
ae,
Viola
Duppel
a,
Kathrin
Müller
a,
Ulrich
Starke
a and
Bettina V.
Lotsch
*abf
ab,
Leslie M.
Schoop
c,
Daniel
Wurmbrand
d,
Sourav
Laha
a,
Filip
Podjaski
ae,
Viola
Duppel
a,
Kathrin
Müller
a,
Ulrich
Starke
a and
Bettina V.
Lotsch
*abf
aMax Planck Institute for Solid State Research, Heisenbergstrasse 1, 70569 Stuttgart, Germany. E-mail: b.lotsch@fkf.mpg.de
bLudwig-Maximilians-Universität München, Butenandtstrasse 5-13, 81377 Munich, Germany
cPrinceton University, Princeton, USA
dUniversity of Konstanz, Konstanz, Germany
eEcole Polytechnique Fédérale de Lausanne, Station 12, 1015 Lausanne, Switzerland
fNanosystems Initiative Munich (NIM) and Center for Nanoscience (CeNS), Schellingstrasse 4, 80799 Munich, Germany
First published on 6th October 2018
Abstract
In solids, heterogeneous catalysis is inherently bound to reactions on the surface. Yet, atomically efficient preparation of specific surfaces and the characterization of their properties are impeding its applications towards a clean energy future. Here, we present the synthesis of single layered IrOOH nanosheets and investigations of their structure as well as their electrochemical properties towards oxygen evolution under aqueous acidic conditions. The nanosheets are synthesized by treating bulk IrOOH with a tetrabutylammonium hydroxide solution and subsequent washing. Electron diffraction shows that the triangular arrangement of the edge sharing Ir(O,OH)6 octahedra found in the layers of bulk IrOOH is retained after exfoliation into single layers. When incorporated as an active component in Ti electrodes, the nanosheets exhibit a Tafel slope of 58(3) mV dec−1 and an overpotential of η10 mA cm−2 = 344(7) mV in 0.1 M HClO4, while retaining the trivalent oxidation state of iridium. They outperform bulk rutile-IrO2 and bulk IrOOH as electrocatalytic water oxidation catalysts under the same conditions. The results of this study on the structure–property relationships of low valence IrOOH nanosheets offer new pathways for the development of atom efficient, robust and highly active oxygen evolution catalysts.
1 Introduction
Electrochemical splitting of water into hydrogen and oxygen is of immense technological interest to realize hydrogen as a storage medium for the energy generated from renewable resources.1 Even though the generation of hydrogen is the main goal, the oxygen evolution reaction (OER) is the bottleneck in this process.2,3 Due to the number of complex intermediates and the need to transfer four electrons instead of two in a concerted fashion, this reaction is kinetically challenging, leading to a higher overpotential needed to drive the oxidation of water. Apart from these energetic requirements, OER catalysts also have to be cheap to manufacture, stable over time and chemically resistant to environments of high and/or low pH values.4While many newly investigated catalysts meet most of these stringent requirements,5–10 none of them can compete with ruthenium oxide and iridium oxide regarding catalysis in acidic media. Rutile-type IrO2 (rutile-IrO2) is a state-of-the-art catalyst and it is used to benchmark the activities of new materials due to its high stability against decomposition under acidic conditions.11
To keep iridium oxide catalysts competitive despite their very low natural abundance, strategies such as nanostructuring, scaffolding and electronic design have been applied to increase their OER activity whilst minimizing the amount of active catalyst material. The synthesis of nanoparticles shows great promise, as a small overpotential of η = 0.25 V generates a current density of 4 μA cmox−2 in rutile-IrO2 nanoparticles with a diameter of 7 nm.12 Yet, at this diameter, the nanoparticles consist of only a few unit cells, leaving little space for further size reduction. While other nanostructures such as nanorods,13 thin films14 and mesoporous IrO2 (ref. 15) were explored, none of them have yielded atomically thin catalysts. Other highly relevant investigations are focused on IrOx, an amorphous catalyst containing mixed valence iridium octahedrally coordinated by oxide, hydroxide and water ligands.16–19 The presence of trivalent iridium and the high surface area of the amorphous state are both prerequisites for excellent OER activity, which surpasses that of rutile-IrO2.
In two more recent papers, the promising electrocatalytic properties of IrO2 nanosheets have been explored.20,21 The investigated material is layered and crystalline and acts as a catalyst towards OER in 0.1 M HClO4 with a Tafel slope of 57–62 mV dec−1. Due to the lack of other electrochemical parameters, e.g. the overpotential, as well as the crystal structure of the material, no structure–property–activity relationships were established.
Here, we present IrOOH nanosheets prepared from recently investigated bulk IrOOH via the tetrabutylammonium hydroxide (TBAOH) exfoliation route. The results of our investigation link the structure of the nanosheets and their electrochemical behavior. We studied this system due to the layered nature of the parent compound as well as the rare oxidation state of Ir observed in the bulk material.22 Similar to the bulk compound, the nanosheets consist of a triangular arrangement of IrIII(O,OH)6 octahedra and are stable in acidic environments. Yet, due to an advantageous combination of single layer morphology and a low iridium valence state, the IrOOH nanosheets surpass both bulk IrOOH and rutile-IrO2 as catalysts for the OER under the same conditions.
2 Results and discussion
2.1 Preparation and structure of IrOOH nanosheets
IrOOH nanosheets were prepared by an ultrasonication-assisted exfoliation of bulk IrOOH (obtained by acid treatment of K0.75Na0.25IrO2) in an aqueous TBAOH solution and subsequent washing. The resulting dispersion was deep blue as displayed in the inset in Fig. 1a, similar to the bulk IrOOH powder. The nanosheets were stabilized by a zeta-potential of −36.4 mV, indicating a negatively charged surface as well as an intermediate dispersion stability versus agglomeration. Dispersions of the monolayers were stable against agglomeration for 4–6 weeks.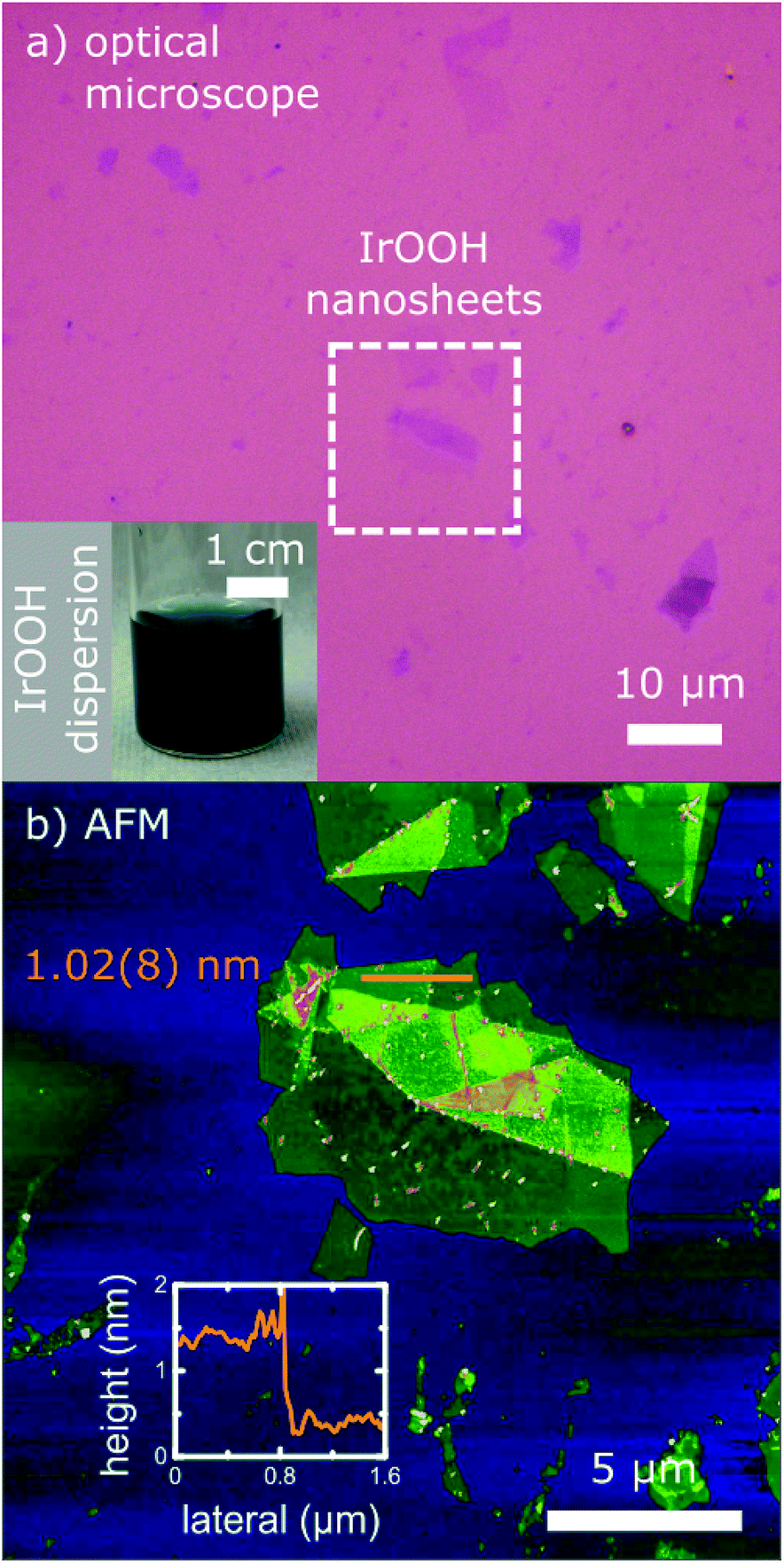 | ||
| Fig. 1 (a) Optical microscope and (b) AFM image of the IrOOH nanosheets; the inset of (a) shows the dark blue aqueous dispersion of the nanosheets; inset of (b) displays an exemplary height profile with an average height value of a single layer of IrOOH measured on a SiO2 (270 nm)–Si substrate. | ||
The IrOOH nanosheets could easily be deposited on various surfaces, either on SiO2–Si substrates (270 nm SiO2, thermally grown) as displayed in Fig. 1 or on Ti substrates (Fig. S3†). After drying, no redispersion of the deposited layers was observed upon wetting with water, which indicates the absence of TBA+ and its hydrate shell. The single layers have lateral dimensions between 0.5 and 7 μm and are 1.02(8) nm in height, as can be seen in Fig. 1b. The monolayer height differs from the height of 0.5 nm of a single crystalline slab in the bulk22 due to a layer of water surrounding the nanosheet, which is subsequently trapped between the nanosheet and the underlying support.23
Before washing, a nanosheet layer height of 1.60(8) nm was observed, which suggests the presence of TBA+ between the nanosheet and the substrate. Upon washing, TBA+ is removed and the height reduced from 1.6 to 1.0 nm. The difference coincides with the height of the TBA+ cation. The absence of TBA+ is also indicated by the energy dispersive X-ray spectroscopy (EDX) line scans in transmission electron microscopy (TEM), where no nitrogen was detected.
The in-plane structure of the IrOOH nanosheets was investigated by TEM with selected area electron diffraction (SAED), EDX on single nanosheets and powder X-ray diffraction (PXRD) on a nanosheet pellet. Fig. 2a depicts an exemplary TEM bright field image of several translucent IrOOH nanosheets.22 The translucency towards the electron beam is typical for very thin samples and indicates that the specimen consists of one or a few layers. TEM-EDX measurements showed that the ratio of iridium to oxygen was Ir![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O = 1:2.3 measured on the Ir M-line and O K-line. Considering the high uncertainty in the detection of light elements such as oxygen using the EDX detectors, this ratio is close to the ideal stoichiometry of Ir:O for IrOOH. No nitrogen was detected, indicating the absence of TBA+. We therefore assume that charge neutrality is attained by protonation of the [IrO2]− backbone. SAED revealed a hexagonal diffraction pattern, which was compared to a simulation based on the crystallographic data of bulk IrOOH as depicted in Fig. 2b and c. The reflections were indexed based on the bulk IrOOH structure with the d-values d(100) = 2.71 Å, d(110) = 1.56 Å, d(020) = 1.35 Å, d(210) = 1.02 Å and d(030) = 0.90 Å.22
:O = 1:2.3 measured on the Ir M-line and O K-line. Considering the high uncertainty in the detection of light elements such as oxygen using the EDX detectors, this ratio is close to the ideal stoichiometry of Ir:O for IrOOH. No nitrogen was detected, indicating the absence of TBA+. We therefore assume that charge neutrality is attained by protonation of the [IrO2]− backbone. SAED revealed a hexagonal diffraction pattern, which was compared to a simulation based on the crystallographic data of bulk IrOOH as depicted in Fig. 2b and c. The reflections were indexed based on the bulk IrOOH structure with the d-values d(100) = 2.71 Å, d(110) = 1.56 Å, d(020) = 1.35 Å, d(210) = 1.02 Å and d(030) = 0.90 Å.22
 | ||
Fig. 2 (a) TEM image and (b) SAED pattern of the IrOOH nanosheets, (c) simulated SAED pattern based on the crystallographic data from bulk IrOOH in the space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 along [001] zone axis; (d) TEM image and (e) SAED pattern of the IrOOH nanosheets after electrocatalysis; (f) PXRD pattern of the IrOOH nanosheet pellet; the small and broad peaks at 2θ = 22° and 25° probably arise from the (111), (201) and (112) reflections, indicating some degree of order along the c-direction in the restacked nanosheet pellet. m1 along [001] zone axis; (d) TEM image and (e) SAED pattern of the IrOOH nanosheets after electrocatalysis; (f) PXRD pattern of the IrOOH nanosheet pellet; the small and broad peaks at 2θ = 22° and 25° probably arise from the (111), (201) and (112) reflections, indicating some degree of order along the c-direction in the restacked nanosheet pellet. | ||
In order to verify the structural integrity of the IrOOH nanosheets under anodic conditions, we analyzed a multilayer nanosheet film by TEM imaging and SAED after electrocatalysis, which resulted in the image and diffraction pattern displayed in Fig. 2d and e. The IrOOH nanosheet film catalyzed the OER under chronopotentiometric conditions at j = 1 mA cm−2 in 0.1 M HClO4 for 7 h and was mechanically removed from the Ti substrate prior to analysis. The TEM image of the post OER-catalysis IrOOH nanosheet film reveals a multilayer consisting of several nanosheets, similar to the nanosheet film prior to catalysis as shown in Fig. S3.† The SAED pattern shows rings of reflections typical for multilayers of nanosheets which form during the creation of the electrodes on the Ti substrate. The observed d-values of d(100) = 2.72 Å and d(110) = 1.57 Å indicate the retention of the in-plane crystal structure after catalysis. Thus, the retention of the nanosheet morphology as well as the crystal structure highlights the extraordinary chemical stability of IrOOH under anodic conditions.
PXRD on a pellet of the nanosheet in transmission geometry yields the in-plane diffraction patterns shown in Fig. 2f. The structural model with the lattice parameters a = 3.136 Å and c = 91.61 Å was fitted to the diffraction patterns using the LeBail method. The turbostratic disorder introduced by restacking was simulated by the expanded c-lattice parameter.24,25 The reflections with the main intensity were indexed based on the structure of bulk IrOOH and were identified as d(100) = 2.72 Å, d(110) = 1.57 Å, and d(020) = 1.36 Å. The small, broad reflections at 2θ = 22° and 25° indicate the introduction of some degree of order during the restacking of the nanosheets.
A comparison of the diffraction data of the nanosheets and known layered bulk compounds is listed in Table 1. The results from the SAED measurements of the nanosheets only slightly deviate from the values obtained from TEM and X-ray diffraction of the bulk compounds. As the deviations are well within the margin of error of electron diffraction (≈5%) and TEM-EDX suggests a composition close to the formula IrO2, the data indicate the retention of the in-plane structure and composition of IrOOH during the exfoliation process. The results were reproducible for single nanosheets (TEM) as well as an ensemble of nanosheets in the form of a pellet (PXRD). Consequently, the nanosheets dispersed in water consist of a triangular layer of the edge sharing IrO6 octahedra, similar to the nanosheets of [MnO2]x−, [CoO2]x−, and [RuO2]x−,26–28 as well as the structure of a slab of brucite-type Mg(OH)2.
| (hkl) | IrOOH nanosheet TEM, d [nm] | IrOOH nanosheet PXRD, d [nm] | IrOOH bulk TEM, d [nm] | IrOOH bulk PXRD, d [nm] | K0.75Na0.25IrO2 bulk SXRD, d [nm] |
|---|---|---|---|---|---|
| (100) | 0.267 | 0.272 | 0.271 | 0.270 | 0.272 |
| (110) | 0.154 | 0.157 | 0.156 | 0.156 | 0.157 |
| (020) | 0.134 | 0.136 | 0.135 | 0.135 | 0.136 |
| (210) | 0.101 | 0.102 | 0.102 | 0.103 | |
| (030) | 0.089 | 0.090 | 0.090 | 0.091 |
The IrOOH nanosheets are structurally and chemically related to other systems which are relevant to electrocatalysis, as displayed in Fig. 3. The first related material is an electrocatalytically activated Ir–Ni mixed oxide thin film system, which features SAED patterns similar to a layered brucite-type structure with increasing Ni content while being X-ray amorphous.29 Under highly corrosive anodic conditions, Ni is leached out, leaving behind a porous surface with an extraordinarily high hydroxyl surface coverage, as displayed schematically in Fig. 3b and discussed in more detail further below. Other related compounds include the highly active oxyhydroxides of the 3d transition metals Mn, Fe, Co, and Ni as well as the layered double hydroxides.9,30 The structure of a CoOOH layer is shown in Fig. 3c. The oxyhydroxide electrocatalysts exhibit some of the lowest overpotentials with regard to OER catalysis for medium to high pH values. Further enhancement in the activity of these layered catalysts was achieved by exfoliation, doping, or the addition of phosphate or borate buffers.9,31,32 The IrOOH nanosheets also structurally resemble the (2 × 2) iridium oxide overlayer, which grows on the Ir(111) surface under low oxygen pressures at room temperature.33 The structure model that best describes the measured data is displayed in Fig. 3d, where the oxygen atoms reside above or below the vacancy in the triangular top layer of iridium. An Ir–O distance of 2.04 Å has been observed, a value similar to the Ir–O distances of 2.070(5) Å and 2.041(4) Å in K0.75Na0.25IrO2 and bulk IrOOH, respectively, obtained from X-ray diffraction.22
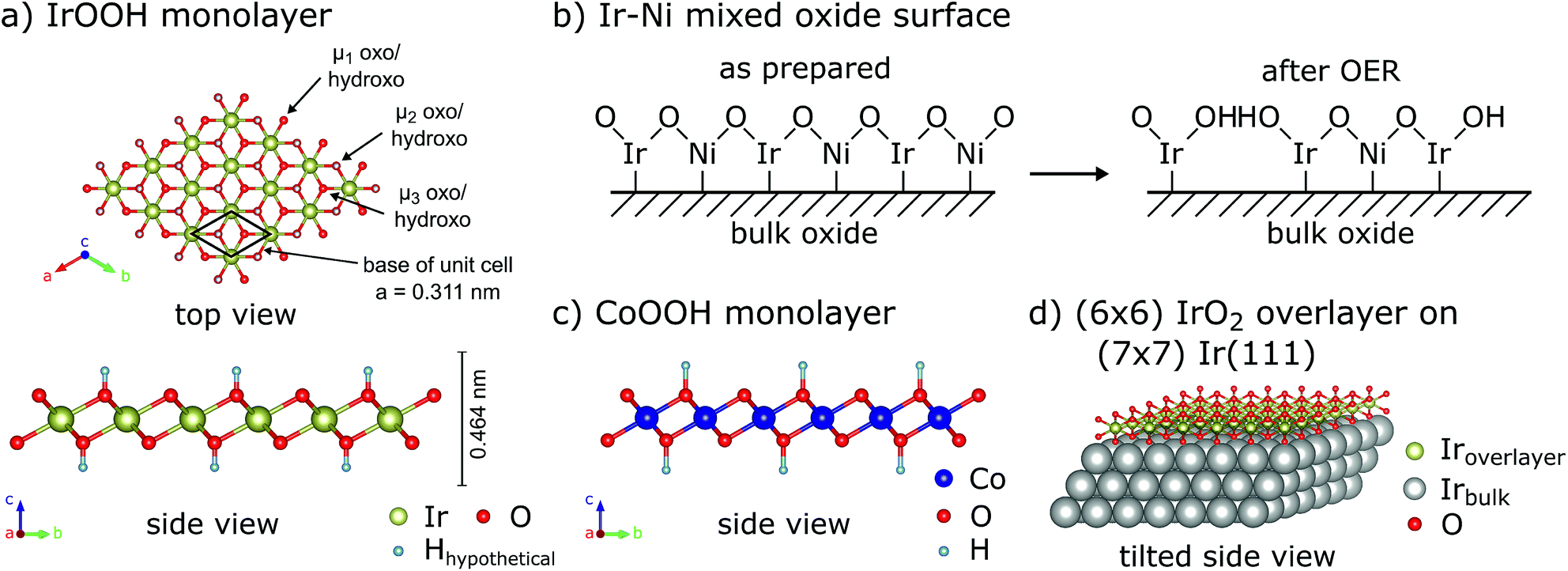 | ||
| Fig. 3 (a) Top and side views of the IrOOH nanosheet structure, μ-oxo/hydroxo-bridges indicated, (b) schematic of acid leaching in Ir–Ni mixed oxide according to Reier et al.,29 (c) side view of a CoOOH monolayer9,30 and (d) tilted side view of the (2 × 2) IrO2 overlayer on Ir(111).33 | ||
Recent work reported on the nanosheets of IrO220 which could have a structure similar to the IrOOH nanosheets presented here. The similar in-plane lattice parameters of 3.1 Å as well as the presence of trigonal/hexagonal symmetry within the plane hint at similarities between the nanosheets of IrO2 and the IrOOH nanosheets prepared by our method. In the following paragraph, we present the relation between the crystal structure and electrochemical properties of the IrOOH nanosheets.
2.2 Electrochemical characterization
To investigate the relationship of the IrOOH structure and its electrochemical properties, the IrOOH nanosheets were incorporated into electrodes by drop-casting the nanosheet dispersion onto titanium. Our deposition approach utilizes the intermediate stability of the IrOOH nanosheet dispersion and the subsequent agglomeration upon evaporation of the solvent. Electrodes were prepared by heating Ti platelets in an aqueous solution of oxalic acid to remove the surface oxide and then drop-casting an ink of the catalyst onto the Ti substrate heated to 160 °C, followed by annealing for 30 min at the same temperature to remove the residual solvent.34 Once the sheets agglomerate on the Ti surface, they do not redisperse, thus eliminating the need for a binder. This is crucial, as possible carbon-based binders have limited stability at the high potentials required during electrolysis in acidic media.35 At least five as-prepared dimensionally stable anodes (DSA) with loadings of 0.2 mg cm−2 and a geometric surface area of 1 cm2 were characterized for the IrOOH nanosheets, bulk IrOOH and rutile-IrO2 in a water splitting setup with an Ag/AgCl/saturated KCl reference electrode, which is schematically depicted in Fig. 4a. The nanosheet coverage of the Ti substrate was estimated to be greater than 95% of the substrate surface from SEM, as only very few spots of the bare Ti substrate were found by back scattered electron detection. The SEM images and additional comments on the electrode coverage are presented in the ESI.† The electrocatalytic properties of the IrOOH nanosheets were investigated by collecting the current–voltage curves of a 0.1 M HClO4 electrolyte. The activities were compared to those of bulk IrOOH and bulk rutile-IrO2 in terms of the Tafel slope, overpotential at 10 mA cm−2 and the current density at 300 mV of overpotential. The current–voltage data were fitted to the Tafel equation (eqn (1)),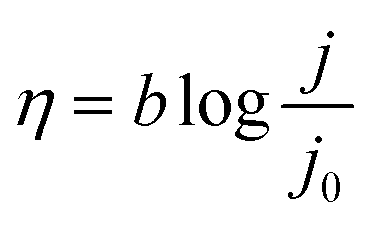 | (1) |
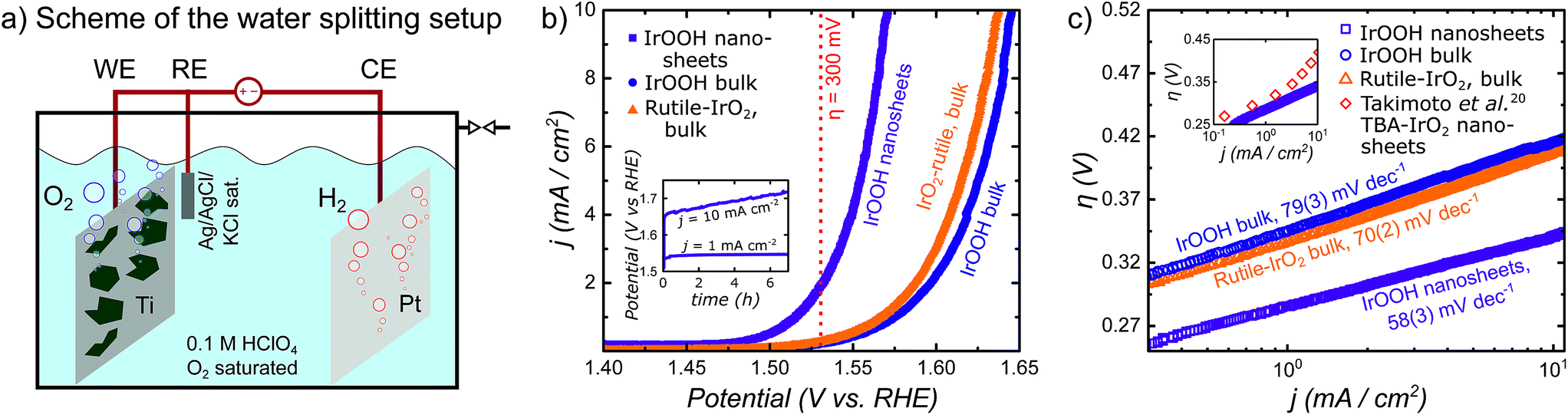 | ||
| Fig. 4 (a) Scheme of the water splitting setup; (b) plot of the current density j vs. the potential with a visual cue for the readout of j at η = 0.3 V; the inset shows chronopotentiometry at j = 1 and 10 mA cm−2; (c) Tafel plot of the overpotential η vs. the logarithmic representation of the current density j and the extracted Tafel slopes; the inset of (c) shows comparison of the IrOOH nanosheets and IrO2 nanosheets reported by Takimoto et al.20 | ||
The Tafel slope was extracted by fitting the Tafel equation to the linear region of the plot of η vs. logj. We found bulk rutile-IrO2 and IrOOH to have Tafel slopes of 70(2) mV dec−1 and 79(3) mV dec−1, respectively, while electrodes modified with IrOOH nanosheets feature a slope of 58(3) mV dec−1. Previously, a Tafel slope of approximately 60 mV dec−1 has been found in the bulk rutile-IrO2 electrocatalysts investigated under acidic conditions. A Tafel slope of this magnitude has been associated with the restructuring process between two energetically inequivalent surface bound hydroxyl groups.36,37 The restructuring of these hydroxyl groups might also be responsible for the rate determining step in bulk IrOOH and nanosheets; however, more careful systematic investigations are needed to elucidate the surface chemistry of IrOOH. As seen in the inset of Fig. 4c, the nanosheets of IrOOH and IrO2 (ref. 20) show similarities in the Tafel slope, thus indicating that the previously reported IrO2-nanosheets feature a similar catalytically active environment.
The overpotential η needed to drive the OER was calculated for a current density of j = 10 mA cm−2 utilizing the Tafel equation. The current density of j = 10 mA cm−2 mimics the output of a high end solar to hydrogen device with 12% efficiency.9 The overpotentials of bulk rutile-IrO2 and IrOOH are η10 mA cm−2 = 415(5) mV and η10 mA cm−2 = 433(5) mV, respectively. The IrOOH nanosheets exhibit a significantly reduced overpotential of η10 mA cm−2 = 344(7) mV. After a small initial increase, the OER potential of the nanosheet electrodes remained constant for j = 1 mA cm−2 over 7 h, as seen in the inset of Fig. 4b. As chronopotentiometry was performed in 0.1 M HClO4, the measurements prove the acid stability of the IrOOH nanosheet electrodes. At a current density of 10 mA cm−2, the intense O2 gas bubble formation led to a ragged chronopotentiometric curve as well as to visually observable catalyst ablation and thus a gradual reduction in activity.
A third descriptor used to compare the performance of the electrocatalysts is the current density j measured at an overpotential of 300 mV. Bulk rutile-IrO2 and IrOOH have relatively low current densities of j300 mV = 0.217(7) mA cm−2 and j300 mV = 0.172(15) mA cm−2, respectively, while the current density of the nanosheet electrodes at η = 300 mV is approximately one order of magnitude larger with j300 mV = 1.8(4) mA cm−2. The high current density observed in the nanosheet electrodes could potentially indicate a higher reaction rate of the nanosheets compared to that of the bulk materials. Thus, exfoliation results in an increased electrocatalytic activity of the nanosheets compared to the bulk compounds, as described by the summary of the three electrocatalytic indicators in Table 2.
| Active electrode material | Tafel slope b [mV dec−1] | η at 10 mA cm−2 [mV] | j at 300 mV [mA cm−2] |
|---|---|---|---|
| IrOOH nanosheets | 58(3) | 344(7) | 1.8(4) |
| IrOOH bulk | 79(3) | 433(5) | 0.172(15) |
| Rutile-IrO2 bulk | 70(2) | 415(5) | 0.217(7) |
2.3 X-ray photoelectron spectroscopy
To verify the retention of the trivalent oxidation state specific to the IrOOH nanosheets, the nanosheet electrodes were characterized by X-ray photoelectron spectroscopy (XPS) after electrolysis and compared to bulk IrOOH and bulk rutile-IrO2. The Ir 4f7/2 and 4f5/2 peaks observed in each spectrum were fitted with two components and additional satellites according to the Wertheim–Kotani model, which states that more than one component is necessary for the fit due to the asymmetric peak shape.38 The peak shapes become less asymmetric when going from rutile-IrO2 to bulk and nanostructured IrOOH, which has been associated with a decrease in metallicity.29 The iridium valence state was analyzed by comparing the position of the Ir 4f7/2 and 4f5/2 peaks, which were observed at 62.3 eV and 65.33 eV in the spectra of the nanosheet electrodes after 60 cycles of cyclic voltammetry at 5 mV s−1 between 1.0 V and 1.5 V (vs. Ag/AgCl). Bulk IrOOH and bulk rutile-IrO2 featured peaks at 62.5 and 65.5 eV as well as 61.8 and 64.8 eV, respectively, with the related spectra and fits depicted in Fig. 5. The 4f iridium binding energies in the nanosheet electrode samples are in almost perfect agreement with the binding energies of bulk IrOOH, indicating that iridium is present as a trivalent species surrounded by oxide/hydroxide ions in the IrOOH nanosheets. Similar XPS results on trivalent iridium with an octahedral oxide/hydroxide coordination have been recently reported for crystalline bulk IrOOH,22 the amorphous OER catalyst IrOx and a predicted trivalent Ir-defect site in rutile-IrO2.19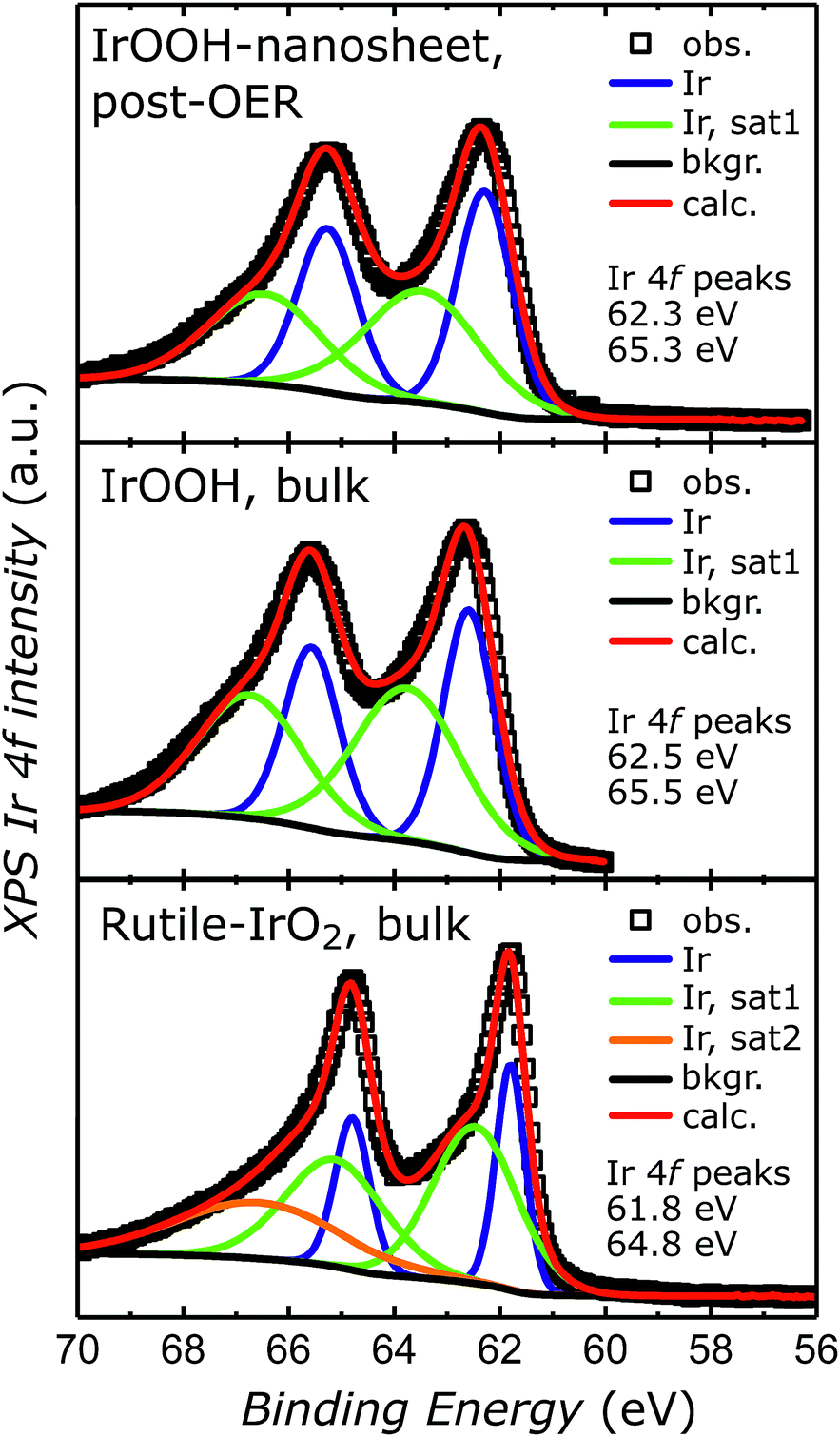 | ||
| Fig. 5 XPS spectra of the IrOOH nanosheet electrode surface after OER, bulk IrOOH and bulk rutile-IrO2 in the energy region of the Ir 4f transition. | ||
2.4 Discussion
In light of these findings, the differences in the electrochemical behavior of the IrOOH nanosheets compared to bulk IrOOH and rutile-IrO2 can be understood by considering the octahedral connectivity, electron configuration of iridium, morphology variations, changes due to the protonation and density of the surface hydroxyl species. In IrOOH, the [IrO6] octahedra are edge connected to six neighbors, while one octahedron is edge linked to two octahedra and corner connected to another eight octahedra in rutile-IrO2. This difference in connectivity could have a sizeable influence on the number of possible configurations as well as the energetic landscape of the active site. Additionally, prior research suggests that tetravalent rutile-IrO2 with its 5d5 electron state is a metallic conductor, while layered IrOOH is a semiconductor with a low spin 5d6 electron configuration for trivalent iridium.22 Such a decrease in valency of the metal center has been correlated with an increased OER activity in mixed valence compounds such as amorphous IrOx19,39,40 or the pyrochlore Y2Ru2O7−δ.41 In both examples, the presence of trivalent metal centers has been identified as a critical ingredient for excellent catalytic activity. In bulk IrOOH, Ir is purely trivalent, which leads to the presence of a band gap. While this represents a disadvantage in terms of conductivity, IrOOH can be exfoliated into nanosheets. In our work, these nanosheets showed a reduced overpotential compared to the bulk of IrOOH and rutile-IrO2. Similar reductions in the OER overpotential of nanosheet materials have been observed in previous investigations on semiconducting layered double hydroxides.9 There, apart from exposing an increased number of active sites, exfoliation leads to a shortened electron path length and, possibly, a more facile percolation from the conducting substrate through the catalyst to the solid–liquid interface,9,42 and consequently to smaller interfacial resistances. Exfoliation can also free up the path towards metallic edge states on undercoordinated metal cations, which have been linked to the enhanced catalytic activity for the hydrogen evolution reactions in MoS2 nanosheets.43–45 Another potentially beneficial factor is good coverage with hydroxyl species on the catalyst surface, which is 50% in IrOOH. In a previous work, this high hydroxyl coverage was created on the surface of thermally grown Ir–Ni mixed oxide films by leaching of Ni under anodic conditions, as shown in Fig. 3b.29 There, the porous surface features a high density of weakly bound hydroxy groups, which act as the reactive surface intermediates for the OER. A combination of these effects could explain the decrease in overpotential observed upon the exfoliation of IrOOH, which allows its use as a catalyst at low precious metal loadings.Additionally, the presented catalyst is interesting when considering the OER mechanism and degradation pathways found in rutile-IrO2 under anodic conditions. Recent literature suggests that the oxidized metallic Ir films and rutile-IrO2 catalyse O2 evolution in a two-electron per Iridium reaction going through IrV and IrIII intermediates.35,46 Degradation occurs either by oxidation of pentavalent Ir oxyhydroxide to water soluble hexavalent IrO42− on the active site or by dissolution of oligomeric IrOx hydrates containing corner sharing, mixed tri- and tetravalent IrO6 octahedra.19,35,39,46 In contrast, the IrOOH nanosheets presented in this work feature a lower valence state of Ir in a motif similar to brucite-type structures such as CoOOH. There, cobalt is trivalent and arranged in layers of edge sharing Co(O,OH)6 octahedra in the resting state. Under OER conditions, the oxidation state of Co cycles between CoII, CoIII and CoIV.47,48 Similarly, the change to trivalent iridium as well as the layered structural motif could shift the balance away from the degradation reactions towards the competing O2 pathway, thus decreasing the likelihood of oxidative decomposition in the nanosheets. Therefore, the IrOOH nanosheets presented in this work are not only of high interest for fundamental studies on electrocatalytic oxygen evolution from water, but also have the potential to excel in industrial applications.
3 Conclusions
We report a procedure for the exfoliation of bulk IrOOH into an aqueous dispersion of IrOOH nanosheets. TEM images along with SAED patterns show that the single layers retain the triangular lattice of the CdI2-type structure of the parent compound IrOOH consisting of edge-sharing IrO6 octahedra. The nanosheets were incorporated into a dimensionally stable anode on titanium and yielded an overpotential of 344 mV at 10 mA cm−2 and a Tafel slope of 58 mV dec−1. Thus, exfoliation leads to materials with increased electrocatalytic activity, which is comparative or even superior to that of bulk IrOOH (η = 433 mV, b = 79 mV dec−1) or that of the state-of-the-art catalyst bulk rutile-IrO2 (η = 415 mV, b = 70 mV dec−1). As chronopotentiometry shows little change in activity and XPS indicates the retention of the trivalent state in iridium after the OER, both methods illuminate the potentially long lifetime of IrOOH nanosheet based catalysts. Thus, the presented material is a promising platform for fundamental studies of low valence iridium as an OER catalyst and could be of great interest for large-scale industrial heterogeneous catalysis.4 Experimental
4.1 Material preparation
:IrOOH of 2:1. After ultrasonicating the dispersion for 30 min, it was centrifuged at 1000 rpm and the supernatant was separated from the unexfoliated solid components. Subsequently, the supernatant dispersion was centrifuged at 15000 rpm for 30 min to precipitate the nanosheets, and the TBAOH solution was decanted and exchanged for doubly distilled H2O, yielding a dark blue dispersion. This exchange of the TBAOH-containing supernatant for water was critical, as the nanosheets deposited from a TBAOH containing solution redispersed after deposition, impeding their electrochemical characterization. To prevent agglomeration, the suspension was stored at 4 °C. The nanosheets were investigated by transmission electron microscopy (TEM) with EDX, atomic force microscopy (AFM), zeta potential and electrochemical measurements. A nanosheet pellet was investigated by transmission powder X-ray diffraction.
Electrodes of the nanosheets and bulk compound IrOOH and commercially available rutile-IrO2 were prepared by drop-casting. The nanosheet suspension was drop-cast onto the etched Ti platelets at 160 °C in steps of 0.1 mL until coverage of 0.2 mg cm−2 was reached. The substrates were then annealed for 20 min at 160 °C. SEM images of the thus covered Ti platelets are displayed in Fig. S3.† The electrodes of bulk IrOOH and commercially available powder of bulk rutile-IrO2 were processed into inks by dispersing 4 mg mL−1 in an isopropanol water solution (2:1, v/v) in an ultrasonic bath for 30 min, then deposited onto the Ti platelets at T = 160 °C and heated for another 20 min. The loaded substrates were contacted to a wire using silver paste and the back side insulated with epoxy glue, yielding electrodes of a 1.00 cm2 geometrical surface area.
4.2 Characterization
For the OER measurements, the system was saturated with O2 for at least 15 min prior to the characterization of the WE. Ohmic drop correction was performed using electrochemical impedance spectroscopy (EIS) as well as the current interrupt method. All data were corrected for internal resistance (iR) using the Ivium Potentiostat software. To identify the region of interest, initial cyclovoltametric (CV) measurements were performed over a range of −0.1 to 1.6 V and the tested electrodes were discarded. The long term stability was tested by chronopotentiometry at 1 mA cm−2 over 7 h and at 10 mA cm−2 over 14 h.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support by the Max Planck Society, the German Science Foundation (DFG), the Nanosystems Initiative Munich (NIM), and the Center for Nanoscience (CeNS). Open Access funding provided by the Max Planck Society.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- M. T. Koper, J. Electroanal. Chem., 2011, 660, 254–260 CrossRef CAS.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- J. Bockris and T. Otagawa, J. Electrochem. Soc., 1984, 131, 290–302 CrossRef CAS.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397–426 CrossRef CAS.
- Z. Lu, W. Xu, W. Zhu, Q. Yang, X. Lei, J. Liu, Y. Li, X. Sun and X. Duan, Chem. Commun., 2014, 50, 6479 RSC.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- G. Li, H. Yu, W. Song, X. Wang, Y. Li, Z. Shao and B. Yi, Int. J. Hydrogen Energy, 2012, 37, 16786–16794 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach and A. Ludwig, et al. , Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- E. Ortel, T. Reier, P. Strasser and R. Kraehnert, Chem. Mater., 2011, 23, 3201–3209 CrossRef CAS.
- M. Hüppauff and B. Lengeler, J. Electrochem. Soc., 1993, 140, 598–602 CrossRef.
- T. Reier, D. Teschner, T. Lunkenbein, A. Bergmann, S. Selve, R. Kraehnert, R. Schlögl and P. Strasser, J. Electrochem. Soc., 2014, 161, F876–F882 CrossRef CAS.
- M. Bernicke, E. Ortel, T. Reier, A. Bergmann, J. Ferreira de Araujo, P. Strasser and R. Kraehnert, ChemSusChem, 2015, 8, 1908–1915 CrossRef CAS PubMed.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, C. Massué, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, M. T. Greiner, J. Allan, M. Hashagen, G. Weinberg, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Surf. Interface Anal., 2016, 48, 261–273 CrossRef CAS.
- D. Takimoto, K. Fukuda, S. Miyasaka, T. Ishida, Y. Ayato, D. Mochizuki, W. Shimizu and W. Sugimoto, Electrocatalysis, 2017, 8, 144–150 CrossRef CAS.
- D. Takimoto, Y. Ayato, D. Mochizuki and W. Sugimoto, Electrochemistry, 2017, 85, 779–783 CrossRef CAS.
- D. Weber, L. Schoop, D. Wurmbrand, J. Nuss, E. Seibel, F. F. Tafti, H. Ji, R. J. Cava, R. E. Dinnebier and B. V. Lotsch, Chem. Mater., 2017, 29, 8338–8345 CrossRef CAS.
- A. Lerf and R. Schoellhorn, Inorg. Chem., 1977, 16, 2950–2956 CrossRef CAS.
- D. Yang and R. Frindt, J. Mater. Res., 1996, 11, 1733–1738 CrossRef CAS.
- K. Szendrei, P. Ganter, O. Sànchez-Sobrado, R. Eger, A. Kuhn and B. V. Lotsch, Adv. Mater., 2015, 27, 6341–6348 CrossRef CAS PubMed.
- Y. Omomo, T. Sasaki, L. Wang and M. Watanabe, J. Am. Chem. Soc., 2003, 125, 3568–3575 CrossRef CAS PubMed.
- T. W. Kim, E.-J. Oh, A.-Y. Jee, S. T. Lim, D. H. Park, M. Lee, S.-H. Hyun, J.-H. Choy and S.-J. Hwang, Chem.–Eur. J., 2009, 15, 10752–10761 CrossRef CAS PubMed.
- W. Sugimoto, H. Iwata, Y. Yasunaga, Y. Murakami and Y. Takasu, Angew. Chem., Int. Ed., 2003, 42, 4092–4096 CrossRef CAS PubMed.
- T. Reier, Z. Pawolek, S. Cherevko, M. Bruns, T. Jones, D. Teschner, S. Selve, A. Bergmann, H. N. Nong, R. Schlögl, K. J. J. Mayrhofer and P. Strasser, J. Am. Chem. Soc., 2015, 137, 13031–13040 CrossRef CAS PubMed.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. García de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- C.-M. Chan and W. H. Weinberg, J. Chem. Phys., 1979, 71, 2788–2792 CrossRef CAS.
- W. Sun, Y. Song, X.-Q. Gong, L.-m. Cao and J. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 820–826 CrossRef CAS PubMed.
- C. Spöri, J. T. H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 ( Angew. Chem. , 2017 , 129 , 6088–6117 ) CrossRef PubMed.
- J.-M. Hu, J.-Q. Zhang and C.-N. Cao, Int. J. Hydrogen Energy, 2004, 29, 791–797 CrossRef CAS.
- M. E. G. Lyons and S. Floquet, Phys. Chem. Chem. Phys., 2011, 13, 5314–5335 RSC.
- J. M. Kahk, C. G. Poll, F. E. Oropeza, J. M. Ablett, D. Céolin, J.-P. Rueff, S. Agrestini, Y. Utsumi, K. D. Tsuei, Y. F. Liao, F. Borgatti, G. Panaccione, A. Regoutz, R. G. Egdell, B. J. Morgan, D. O. Scanlon and D. J. Payne, Phys. Rev. Lett., 2014, 112, 117601 CrossRef CAS PubMed.
- Z. Pavlovic, C. Ranjan, Q. Gao, M. van Gastel and R. SchloÌĝl, ACS Catal., 2016, 6, 8098–8105 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. Mayrhofer, J. Electroanal. Chem., 2016, 774, 102–110 CrossRef CAS.
- J. Kim, P.-C. Shih, K.-C. Tsao, Y.-T. Pan, X. Yin, C.-J. Sun and H. Yang, J. Am. Chem. Soc., 2017, 139, 12076–12083 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- M. V. Bollinger, J. V. Lauritsen, K. W. Jacobsen, J. K. Nørskov, S. Helveg and F. Besenbacher, Phys. Rev. Lett., 2001, 87, 196803 CrossRef CAS PubMed.
- J. V. Lauritsen, J. Kibsgaard, S. Helveg, H. Topsøe, B. S. Clausen, E. Lægsgaard and F. Besenbacher, Nat. Nanotechnol., 2007, 2, 53 CrossRef CAS PubMed.
- O. Kasian, J.-P. Grote, S. Geiger, S. Cherevko and K. J. Mayrhofer, Angew. Chem., Int. Ed., 2018, 57, 2488–2491 ( Angew. Chem. , 2018 , 130 , 2514–2517 ) CrossRef CAS PubMed.
- D. K. Bediako, A. M. Ullman and D. G. Nocera, Catalytic oxygen evolution by cobalt oxido thin films, Springer, 2015, pp. 173–213 Search PubMed.
- J. Li, R. Güttinger, R. Moré, F. Song, W. Wan and G. Patzke, Chem. Soc. Rev., 2017, 46, 6124–6147 RSC.
- M. B. Stevens, L. J. Enman, A. S. Batchellor, M. R. Cosby, A. E. Vise, C. D. Trang and S. W. Boettcher, Chem. Mater., 2016, 29, 120–140 CrossRef.
- J. Moulder, W. Stickle, P. Sobol, K. Bomben and J. Chastain, Physical Electronics Division, Perkin-Elmer, Eden Prairie, 1995, 1992 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional details on experimental procedures, SEM images of the electrodes and electrochemical data on iridium oxide based catalysts. See DOI: 10.1039/c8ta07950a |
| ‡ Present address: Ohio State University, Columbus OH, USA. |
| This journal is © The Royal Society of Chemistry 2018 |