
A deeply rechargeable zinc anode with pomegranate-inspired nanostructure for high-energy aqueous batteries†

Abstract
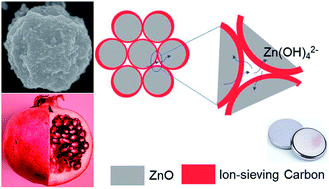
Rechargeable, Zn-based aqueous batteries because of their advantages of inflammability, high energy density, and low material cost are an attractive alternative to lithium-ion and lead-acid batteries for transportation and grid-scale applications. Historically, zinc anodes have shown low utilization and rechargeability in alkaline electrolytes due to the problems of ZnO passivation and Zn(OH)42− dissolution. Herein, we report a nanoscale, pomegranate-structured Zn anode (Zn-pome) fabricated via a bottom-up microemulsion approach to overcome these problems. In the Zn-pome, primary ZnO nanoparticles (ZnO NPs) assemble into secondary clusters after which they are individually encapsulated by a conductive, microporous carbon framework. The small size of ZnO NPs overcomes the issue of passivation, whereas the secondary structure and ion-sieving carbon shell mitigate the dissolution problem. Inductively coupled plasma (ICP) analysis confirms that Zn dissolution from the Zn-pome anode is effectively suppressed, leading to a considerably prolonged cycle life compared to that of a conventional ZnO anode in an alkaline aqueous electrolyte. The Zn-pome anode even maintains the capacity after long resting. This performance is achieved in harsh yet practical conditions: a limited amount of electrolyte, sealed coin cells, and 100% depth of discharge (DOD). This study represents an important step towards producing aqueous, rechargeable, high-energy batteries. In addition, the design principles reported here can be applied to other battery systems involving passivation or dissolution intermediates.
- This article is part of the themed collections: Materials and Nano Research in Atlanta and Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

