
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-standing NASICON-type electrodes with high mass loading for fast-cycling all-phosphate sodium-ion batteries†
Shicheng
Yu
 *a,
Zigeng
Liu
*ab,
Hermann
Tempel
a,
Hans
Kungl
a and
Rüdiger-A.
Eichel
ac
*a,
Zigeng
Liu
*ab,
Hermann
Tempel
a,
Hans
Kungl
a and
Rüdiger-A.
Eichel
ac
aInstitut für Energie- und Klimaforschung (IEK-9: Grundlagen der Elektrochemie), Forschungszentrum Jülich, D-52425 Jülich, Germany. E-mail: zi.liu@fz-juelich.de; s.yu@fz-juelich.de
bMax-Planck-Institut für Chemische Energiekonversion, D-45470 Mülheim an der Ruhr, Germany
cInstitut für Materialien und Prozesse für elektrochemische Energiespeicher- und wandler, RWTH Aachen University, D-52074 Aachen, Germany
First published on 29th August 2018
Abstract
A scalable strategy has been realized to produce anodic NaTi2(PO4)3 and cathodic Na3V2(PO4)3 supported on carbon nanotube fabrics (CNFs) as binder-free, metal current collector-free, carbon additive-free, bendable and self-standing electrodes for sodium-ion batteries. The NaTi2(PO4)3 and Na3V2(PO4)3 particles are not only anchored on the surface of the CNFs but also uniformly embedded in the framework of the CNFs via a two-step coating process followed by annealing treatment. In the relevant voltage range, the high electrochemical stability of the 3D electron conduction network of carbon nanotubes in the self-standing electrodes was confirmed by in situ Raman spectroscopy. Both electrodes possessed a thickness of around 130 μm and a high mass loading of greater than 7.5 mg cm−2 and exhibited a high specific capacity, high rate capability and long lifespan in both half cells and all-phosphate full cells. The all-phosphate full cells delivered more than half of their theoretical capacity even at a high current rate of 100C. Besides, a capacity retention of 75.6% over 4000 cycles at a rate of 20C was achieved. The reason for the capacity fade in the full cell during long-term cycling was the formation of a solid electrolyte interphase layer, as was indicated by XRD, TEM and in operando NMR measurements. Furthermore, the promising practical possibilities of the electrodes and all-phosphate sodium-ion battery were demonstrated by a prototype flexible pouch cell and by stacking multiple electrodes in a laboratory-scale battery.
Introduction
Portable and wearable electronic devices with high flexibility have achieved increasing popularity and a booming market in recent years for a wide range of applications such as roll-up displays, implantable biomedical devices and comfortable electronic skins for health monitoring.1–3 To meet the energy demands of these advanced electronic devices, considerable efforts have been dedicated to the development of flexible energy storage systems with high electrochemical performance. Lithium-ion batteries (LIBs), which combine a high power/energy density and a long lifespan, have been considered for flexible energy storage devices.4 However, as LIBs have entered the fields of electric vehicles and large-scale energy storage systems, a severe bottleneck has appeared owing to the low abundance and relatively high cost of lithium.5,6 Sodium-ion batteries (SIBs) are recognized as alternatives to LIBs because of the abundance, wide geographical distribution and distinctly low cost of sodium sources.7,8On comparing Na with Li, the former has a larger ionic radius and greater atomic mass, which leads to slower insertion/extraction of ions into/from most Na electrode materials than the cases for their Li-containing analogues, such as graphitized carbon materials, silica, transition metal oxides (V2O5), sodium transition metal oxides (NaMO2, M = V, Fe, Mn, Cu, Co, and Ni) and sodium transition metal fluorides (NaMF3, M = Fe, Mn, Ni and V).9,10 However, in some cases Na+-containing host materials have exhibited better electrochemical performance than their Li-containing analogues. In particular, NASICON-type phosphate materials such as anodic NaTi2(PO4)3 (NTP) and cathodic Na3V2(PO4)3 (NVP) both have stable lattice structures and large 3D tunnels for the migration of Na+, which enable zero-strain (de)intercalation during cycling and fast transport of Na+ ions, leading to a high rate capability and long lifespan when used in SIBs.11,12 Specifically, the presence of redox-variable titanium/vanadium ions endows NTP and NVP with a substantial diffusion coefficient for Na+ and a perceptible electronic conductivity. The intrinsic electronic conductivity of these materials is not enough to provide sufficient electronic connectivity among the active particles in the electrodes. To mitigate this issue, doping with different transition metal ions, decreasing the particle size and surface modification in terms of coating with carbon are typical approaches for increasing the electronic conductivity of these materials.12–14
In order to enhance the mechanical flexibility of SIBs, inorganic nano/microsized powders mixed with polymer binders are normally used to prepare the electrodes. However, this method often causes an insufficient specific energy density and poor electrochemical performance because of the low mass loading ratio of the active materials and/or high contact resistance at interparticle junctions.15,16 Hence, the design and fabrication of flexible and binder-free electrodes with a high specific energy density and flexibility in novel geometric designs but without traditional binder, conductive carbon or metal foil current collectors recently have attracted much attention.15–20 Normally, such electrodes are composed of active materials and graphene, carbon nanotubes (CNTs) or carbon nanotube fabrics (CNFs) and feature fast in-plane electrical conductivity, a large specific surface area, an excellent tensile modulus, and high mechanical durability.21–23 The elastic nature of carbon fabric materials also accommodates variations in the volume of embedded electrode materials during cycling and thereby minimizes the degradation of electrode materials due to the associated strain.24,25 From the viewpoint of the large-scale practical application of carbon materials in flexible electrodes, the lower market price and sufficient content of graphitized carbon are the advantages of CNT/CNF materials in comparison with graphene. However, from the aspect of electrode preparation, the direct growth of electrode materials on the surface of CNT materials usually results in a thin layer with a low loading of active materials, unsatisfactory tap density, and reduced energy and power density. Although a thick layer of electrode materials can be deposited on the surface of CNT substrates, this thick layer tends to detach from the CNT substrates during cycling, which leads to serious capacity fade.26,27 Therefore, for practical applications, the fabrication of CNT/CNF-supported electrodes with a large mass loading, high rate capability, high flexibility and a long lifespan remains a challenge.
Here, we report a powerful but easy approach for preparing binder-free self-standing NASICON-type electrodes, namely, a NaTi2(PO4)3/CNFs (NTP/CNFs) anode and a Na3V2(PO4)3/CNFs (NVP/CNFs) cathode, with efficient electronic/ionic conductivity and a high mass loading. For both electrodes, submicrometer-sized active materials were directly grown on the surface of CNFs and, on the other hand, nanosized NTP and NVP were uniformly distributed in bulk CNFs. The efficient interconnected conductive carbon nanotube network and uniformly distributed small NTP and NVP particles enabled effective electronic charge transport, short diffusion paths for Na+ and large spaces for variations in volume, which led to outstanding electrochemical performance of the electrodes in SIBs. In situ Raman and in operando NMR spectroscopy and post-test measurements confirmed the high electrochemical stability of the CNFs and active materials over long-term cycling. In addition, the high electrochemical performance of flexible pouch cells and all-phosphate SIBs assembled with multiple self-standing electrodes demonstrated the impressive quality and practical possibilities of this approach.
Experimental section
Electrode preparation
A4-sized CNFs with a thickness of 115 μm were obtained from Tortech Nano-Fibers Ltd. (Israel). The CNFs were cleaned with concentrated hydrochloric acid and a mixture of ethanol/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, vol/vol) several times before use in order to remove surface impurities. All other chemical reagents (of analytical grade) were purchased from Sigma-Aldrich and used as received.
:1, vol/vol) several times before use in order to remove surface impurities. All other chemical reagents (of analytical grade) were purchased from Sigma-Aldrich and used as received.
A Na–Ti–P–O precursor solution was prepared by the dropwise addition of titanium(IV) isopropoxide (∼0.05 mol) to ammonium hydroxide (30 mL) under constant vigorous stirring until a white gelatinous precipitate formed. The precipitate was washed with 500 mL deionized water to remove excess base and then dissolved in a 1 M solution of oxalic acid (200 mL) at 60 °C under stirring until a transparent solution containing H2[TiO(C2O4)2] was formed. Sodium acetate (5% stoichiometric excess) and a stoichiometric amount of ammonium dihydrogen phosphate were dissolved in water separately and then added slowly to the transparent solution.
A Na–V–P–O precursor solution was prepared by first mixing 0.05 mol ammonium metavanadate and oxalic acid in a stoichiometric ratio of 1:3 in 200 mL deionized water. After magnetic stirring at 50 °C for 12 hours, a dark blue solution was formed. Subsequently, stoichiometric amounts of sodium acetate and ammonium dihydrogen phosphate were dissolved in water separately and then added slowly to the dark blue solution under continuous stirring. Both of the prepared precursor solutions were continuously stirred at 80 °C for 6 hours before use. Oxalic acid in both cases served as a chelating agent during preparation and as a carbon source during the annealing process.
The pretreated CNFs were cut into 8 cm × 8 cm squares and added to the respective prepared precursor solutions. After refluxing at 120 °C overnight, the CNFs were taken out and dried at 50 °C in a vacuum oven overnight to form NTP/CNFs and NVP/CNFs solid precursors. Afterwards, to increase the mass loading, the precursor solutions were uniformly dropped onto the respective solid precursors in appropriate amounts (∼0.5 mL cm−2). After slow removal of the solvent, the solid precursors were dried at 100 °C under vacuum and annealed at 800 °C for 8 hours in N2 atmosphere. NTP/CNFs and NVP/CNFs electrodes were obtained after the removal of the loosely bound powders on their surfaces and uniaxially pressed at 30 MPa. The thickness of the prepared electrodes was around 130 μm, and the mass loading of both electrodes was greater than 7.5 mg cm−2. In addition, the loading of the active materials on the CNFs was tuned by controlling the drop-coating process.
Structural characterization
Analysis of the crystal structure and phase was carried out using an Empyrean (PANalytical, Netherlands) X-ray diffractometer with Cu Kα radiation operated at 40 kV and 40 mA at a scanning rate of 1° min−1. Scanning electron microscopy (SEM) images and EDX maps were recorded using a Quanta FEG 650 (FEI, USA) microscope equipped with an EDAX-TSL detector. For transmission electron microscopy (TEM) investigations, thin sections of the electrodes with a thickness of less than 100 nm were cut using a Leica EM UC6 Ultracut ultramicrotome equipped with a glass knife after the samples were embedded in an epoxy resin embedding kit (Sigma-Aldrich). The sections after cutting were dispensed onto a copper grid. TEM and high-resolution TEM (HRTEM) measurements were conducted on the prepared samples using a Tecnai F20 (FEI, USA) microscope at an accelerating voltage of 200 kV. Thermogravimetric analysis (TGA) was performed using a NETZSCH TGA/STA-QMS 403D thermoanalyzer (Germany) between 30 °C and 700 °C at a heating rate of 2 °C min−1 under an O2 flow. The specific surface area was determined via Brunauer–Emmett–Teller (BET) measurements by N2 adsorption, and a QUADRASORB evo automatic adsorption analyzer (Quantachrome Instruments, USA) was used. The CNFs and prepared electrodes were cut into small pieces and subsequently placed separately in 9 mm BET cells. The mass of the sample in each cell was about 0.1 g. The cells filled with samples were degassed under vacuum with a FlowVac degasser (Quantachrome Instruments, USA) at 180 °C for at least 20 hours prior to data collection. The reproducibility and accuracy of the results for the specific surface area of the samples was ±0.4 m2 g−1, which was confirmed with different samples. Raman spectra were recorded using a SENTERRA Raman microscope (Bruker, Germany) with an excitation wavelength of 532 nm and a 50× objective lens with a long working distance. An in situ Raman cell (ECC-Opto-Std, EL-CELL, Germany) was used to record Raman spectra and perform electrochemical measurements at the same time. At intervals of 30 minutes, acquisition of each Raman spectrum was carried out during charging–discharging of the full cell in the potential range of 0.5–2.0 V at a current rate of ∼0.08C. The laser exposure time was 30 s, with double accumulations for one spectrum, and the size of the confocal pinhole was set at 50 μm. 23Na solid-state nuclear magnetic resonance (23Na ssNMR) and in operando23Na ssNMR spectra were acquired using a Bruker AVIII-400 spectrometer. All the 23Na NMR experiments were carried out using a single-resonance homebuilt in operando probe with a saddle coil with a diameter of 15 mm.28 The 23Na NMR spectra were acquired using a Hahn echo pulse sequence with a 90° pulse length of 9 μs and a recycle delay of 1 s. All the chemical shifts of 23Na were referenced to a 1 M aqueous solution of NaCl (0 ppm). A total of 32 scans were performed for each spectrum in in operando NMR experiment when the full cell was cycling in the potential range of 0.5–2.0 V at a current rate of ∼1C in the initial and final cycles. Between these two in operando NMR measurements, the in operando NMR battery was cycled 1000 times at a constant current rate of ∼10C.Cell assembly and electrochemical measurements
The electrochemical properties of the prepared electrodes were assessed in Swagelok cells. The assembly of all the test cells was carried out in an argon-filled glovebox, where the concentrations of water and oxygen were kept at less than 0.1 ppm. A glass fiber membrane served as the separator, and 1 M NaPF6 in a mixture of ethylene carbonate (EC)/propylene carbonate (PC) (1:1, vol/vol) was used as the electrolyte.
The electrochemical properties of the prepared electrodes were initially investigated in half cells. The binder-free metal substrate-free NTP/CNFs and NVP/CNFs electrodes were cut into disks with a size of 1.13 cm2 and used directly as the working electrode. Metallic sodium foil was used as both the counter and the reference electrode in the half cell experiments. In full cells, the disks of NTP/CNFs and NVP/CNFs were used as the anode and cathode, respectively. The NTP/CNFs electrode was connected to the side of the reference and counter electrodes in all the electrochemical tests performed on full cells. To ensure efficient utilization of the anode material, the value of the capacity balance was selected to be close to 1:1.2 (anode: cathode). Therefore, the area of the anode was 0.785 cm2 and the area of the cathode was 1.13 cm2, and the theoretical capacity of the cell was 0.83 mA h according to the mass of the anode. Furthermore, the capacity of a cell can be increased linearly when multiple electrode pairs are stacked in one cell. In the full cells assembled for in situ Raman measurements, the electrode used for Raman focusing was cut into disks with a size of 3.14 cm2 and pressed onto a copper mesh. The area of the counter electrode was 0.785 cm2. The configuration of the battery case used for the measurements was similar to that used in previous reports.29,30 For the full cells assembled for in operando ssNMR measurements, an in operando NMR battery designed in a similar way to a standard Swagelok cell was used,28 which allowed the geometry of the electrodes to be the same as that in the full cells in the electrochemical measurements. The differences from standard Swagelok cells were that (i) no spring was used and (ii) the area of electrical contact was much smaller (∼0.008 cm2) in the in operando NMR battery.
Cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and galvanostatic charge/discharge measurements for all the cells were conducted using a VMP3 potentiostat (Bio-Logic, France). An MKF120 climate chamber (Binder, Germany) was used to control the temperature during the electrochemical measurements.
Results and discussion
The composition, structure and mass loading of the as prepared NTP/CNFs and NVP/CNFs electrodes were analyzed using XRD, Raman and TGA measurements. The detailed results and discussion are shown in the ESI (Fig. S1 and S2†). On the basis of the TGA results (Fig. S1†), about 23.5 and 27.4 wt% carbon phases were present in NTP/CNFs and NVP/CNFs, respectively, including CNFs and amorphous carbon created by carbonization of the starting materials (mainly oxalic acid). Correspondingly, high mass loadings of 8.02 and 7.63 mg cm−2 for NTP/CNFs and NVP/CNFs were achieved. These values are higher than most of those reported for self-standing electrodes.20,31–33 Hence, these high mass loadings of the electrodes were able to guarantee the high energy density of the SIBs.The surface morphological features of the CNFs before and after washing, as well as those of the NTP/CNFs and NVP/CNFs electrodes, were investigated by SEM. As shown in Fig. 1a and b, the as-received CNFs exhibited overall surface bundling owing to contact effects during fabrication.19 In the SEM images of the washed CNFs (Fig. 1c and d), the CNFs consist of a web of curved continuous nanotubes with a width of 20–30 nm. As shown in Fig. 1e–h, the self-standing NTP/CNFs and NVP/CNFs electrodes exhibited a uniform particle size of ∼200 nm for both materials (NTP and NVP), which were homogeneously distributed on the surface of the 3D network of CNTs. This morphology could effectively improve electron transfer among the particles of the active material on the surface of CNFs.
 | ||
| Fig. 1 SEM images of CNFs as received ((a) and (b)) and after washing with HCl and an ethanol/water (1:1, vol/vol) mixture ((c) and (d)), NaTi2(PO4)3/CNFs ((e) and (f)) and Na3V2(PO4)3/CNFs ((g) and (h)). | ||
Individual decoration of large active particles on the surface of CNFs, as observed in the SEM images, cannot ensure high loadings of the active material on the electrodes. The electrode materials were also located inside CNFs. As evidence, a high-resolution SEM image and the corresponding EDX elemental maps of C, Na, V, P and O are shown in Fig. S3† and were obtained by focusing on a single large NVP particle and its surroundings. As shown in the figure, the characteristic elements in NVP (Na, V, P, and O) were distributed over the entire area of focus, which indicated the presence of active materials between the carbon nanotubes all over the shallow surface of the CNFs. Further insights into the morphology of the bulk electrodes were obtained by TEM observations of thin sections of the electrodes, in which active particles in bulk CNFs are visible. As shown in Fig. 2, the NTP/CNFs and NVP/CNFs electrodes displayed high similarity in terms of the bulk microstructure, as NTP and NVP particles with sizes of less than 100 nm were uniformly embedded in bulk CNFs. The CNTs acted as electron pathways that connected NTP or NVP particles, which endowed the electrode with high electronic conductivity (Fig. 2c and g). Furthermore, the carbon layers on the active materials, as shown in Fig. 2d and h, were mainly graphitized carbon, which improved electron transfer around the active materials. The lattice fringes of NTP and NVP are clearly observed in Fig. 2i and j and indicate their high crystallinity. In these figures the lattice fringes of crystalline carbon layers can be detected, which shows the integrity of CNFs after the synthesis of the active materials. The interlayer spacings were, irrespective of surrounding grains, ∼0.35 nm for NTP/CNFs ((1) and (2) in Fig. 2i) and ∼0.37 nm for NVP/CNFs ((3) and (4) in Fig. 2j).
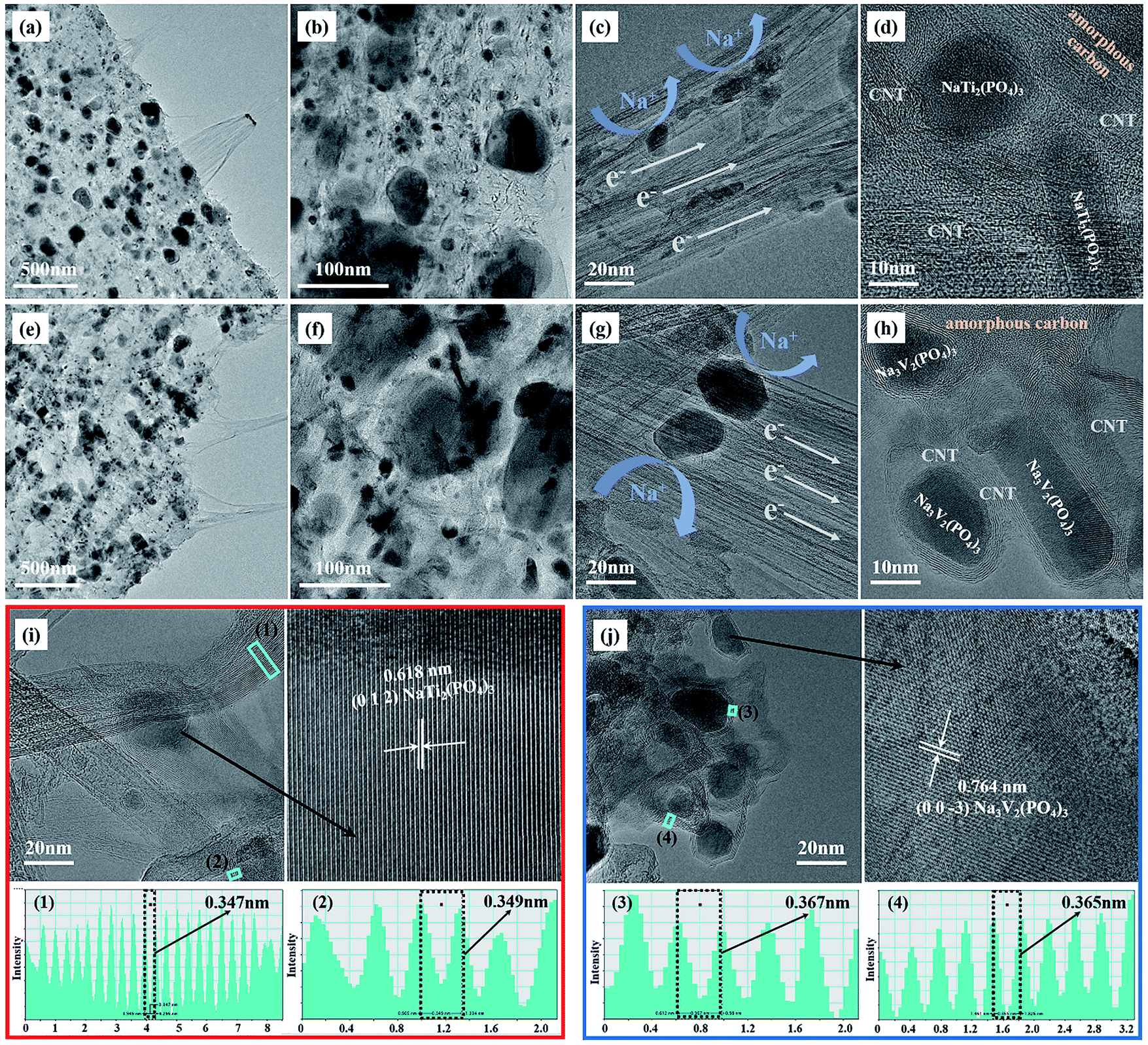 | ||
| Fig. 2 TEM and HR-TEM images of NaTi2(PO4)3/CNFs ((a)–(d) and (i)) and Na3V2(PO4)3/CNFs ((e)–(h) and (j)). The inserted figures ((1)–(4)) in (i) and (j) are the intensity line profiles of graphitized carbon in electrodes for calculating the interlayer spacing. | ||
The full use of the space in the CNFs for loading the active material in such a way that particles of the active material were located on the surface and in the bulk of CNT bunches was proven by SEM and TEM. The formation of the microstructure is shown schematically in Fig. 3. Firstly, hydrophilic NTP or NVP colloids with an anionic (COO−, introduced by oxalic acid) surface were homogeneously distributed in the precursor solution. Secondly, the pretreated CNFs were soaked in the prepared solutions at 120 °C to allow the colloids to penetrate into the CNT bunches via a dip-coating process. Thirdly, the CNFs loaded with the precursors were slowly dried under vacuum and subsequently drop-coated with the prepared liquid precursors (drop-coating process) in order to load more colloids onto the surface of the CNFs. Finally, the surface colloids and the colloids in the bulk CNFs formed nuclei for NTP or NVP crystals and continuously grew during the annealing process at high temperatures. Owing to the difference in the carbon surrounding environment, significant differences were observed between the surface and bulk particles. As can be seen in the TEM images in Fig. 3, the surface particles exhibited a size of around 200 nm with a thin amorphous carbon coating (thickness of ∼3 nm), whereas the bulk particles were embedded in graphitized carbon with particle sizes of less than 100 nm. The formation of the graphitized carbon layer and the small size of the active particles in the bulk CNFs can be explained as follows. The solid colloids of NTP and NVP were first incorporated into CNTs during refluxing. The side products during the crystallization of NTP and NVP evaporated into the CNFs and formed amorphous carbon after calcination at 800 °C. The remaining surface layers of active particles were graphitized carbon, and the particle size was smaller in comparison with the surface particles owing to stress inside the CNFs, which constrained the growth of grains. In contrast, the particles on the surface of the CNFs grew under negligible stress from their surrounding environment, and the coated thin amorphous carbon layers were formed by the carbonization of reactants. Because the parameters of both coating processes were controlled, the agglomeration of particles to form large lumps in the free-standing electrodes or on the surfaces was not observed, which ensured sufficient contact between the liquid electrolyte and the active materials. In order to gain an insight into the ratio of the contents of surface and bulk particles, reference experiments were performed by annealing the CNFs immediately after dip-coating, because the amount of surface particles formed during this step was negligible. The resulting mass loadings of the reference self-supporting films were 3.92 mg cm−2 and 3.77 mg cm−2 for the NTP/CNFs anode and NVP/CNFs cathode, respectively, which were half the values after two-step coating. Hence, the NTP/CNFs and NVP/CNFs electrodes contained surface and bulk particles in a weight ratio of roughly 1:1. The morphological features of the as-prepared electrodes could effectively benefit their electrochemical performance because (i) the full use of the space in the CNFs for mass loading in such a way that particles of the active material are located on the surface and in the bulk of CNT bunches results in a large specific capacity of the electrodes; (ii) the carbon phases can help to circumvent the intrinsic poor electronic conductivity of phosphate electrode materials and improve the binding between the active materials and CNFs;34–37 (iii) the carbon layers can protect the NTP and NVP particles against erosion by the electrolyte and thus increase the cycling life of the electrodes; (iv) the small particle size of the NTP and NVP materials guarantees short diffusion pathways for Na+ in the active materials, as well as relatively large specific surface areas of the electrodes (50.5 m2 g−1 for NTP/CNFs and 49.4 m2 g−1 for NVP/CNFs), which provides excellent kinetics for the intercalation/deintercalation of Na+ and reduces polarization during cycling;38–42 and (v) the high porosity of the electrodes, which was about 69% for the NTP/CNFs and NVP/CNFs electrodes as calculated from the relative and theoretical densities of the materials, could reduce the wetting issue of the electrolyte. Therefore, the as-prepared NTP/CNFs and NVP/CNFs electrodes are expected to be capable of providing high flexibility, high rate capability and stable cycling performance for SIBs in practical applications.
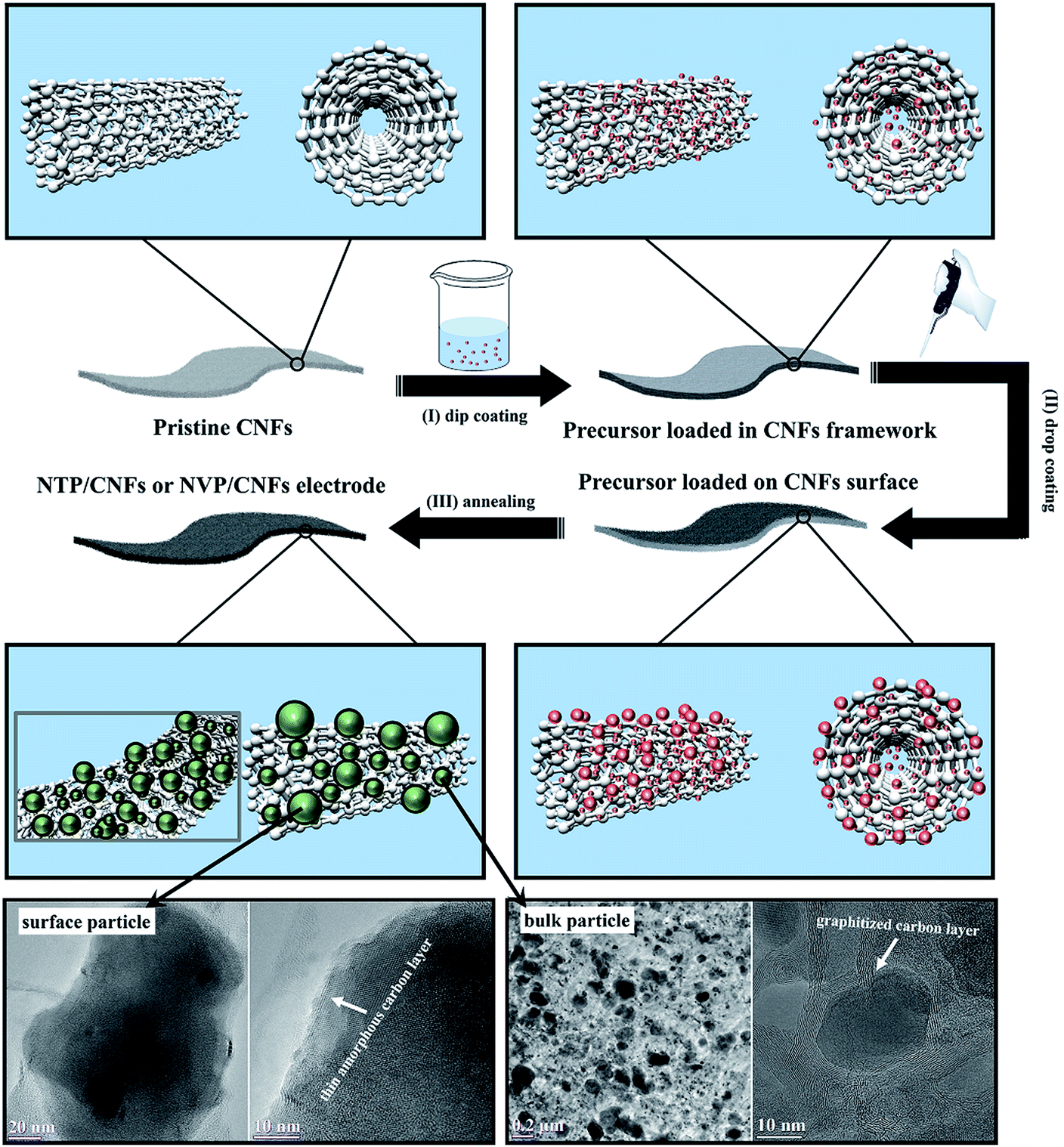 | ||
| Fig. 3 Schematic illustration of the preparation process of self-standing NaTi2(PO4)3/CNFs and Na3V2(PO4)3/CNFs electrodes. The TEM images in the insets were recorded for NaTi2(PO4)3/CNFs and reveal the morphology of surface and bulk particles on/in CNFs. | ||
To assess the Na+ intercalation/deintercalation behavior of the prepared electrodes, cyclic voltammetry (CV) tests were performed in half cells with pretreated CNFs and NTP/CNFs and NVP/CNFs electrodes (Fig. S4a†). In the CV profile of CNFs from 0.001 to 4.0 V vs. Na/Na+, a major reduction peak towards the lower limit of the voltage range was observed, which can be assigned to the intercalation of sodium.43 Except for polarization, which started at 3.53 V and became severe when the voltage was higher than 3.65 V owing to side reactions on the CNT surface, no other redox peaks could be observed in the CV curve when the voltage was higher than 0.5 V. In the voltage ranges corresponding to the phase transition of NTP (1.5–3.0 V vs. Na/Na+) and NVP (2.5–4.0 V vs. Na/Na+), two pairs of redox peaks at 2.20/1.96 V (NTP/CNFs anode material) and 3.44/3.31 V (NVP/CNFs cathode material), respectively, were observed in the CV curves (Fig. S4b†), which corresponded to the Ti(IV)/Ti(III) and V(IV)/V(III) redox couples, respectively.44,45 The overpotentials of the NTP/CNFs anode and NVP/CNFs cathode were 0.24 V and 0.13 V, respectively, which were slightly higher than those in previous reports18,46 owing to the influence of the high scan rate. In addition, a shoulder appeared at 3.53 V during the oxidation process owing to side reactions on the CNT surface in the NVP/CNFs electrode, as in the previous case of the CNFs half cell. Hence, to minimize possible changes in the surface of CNFs and ensure a complete phase transition of NVP at high C-rates, the upper cut-off voltage for cycling NVP/CNFs half cells was set at 3.6 V vs. Na/Na+.
The electrochemical characteristics of the electrodes were further assessed in half cells in the voltage ranges of 1.5–3.0 V (vs. Na/Na+) for NTP/CNFs and 2.6–3.6 V (vs. Na/Na+) for NVP/CNFs. Fig. 4a and b show the charge–discharge profiles of the half cells at 0.05C for the five formation cycles, and it can be observed that the coulombic efficiency was close to 100% for both half cells, which indicated the high reversibility of sodiation/desodiation in the electrodes. Because the current rate was low, both half cells exhibited low charge–discharge overpotentials in comparison with their CV results. The profiles of the NTP/CNFs half cell displayed charge and discharge voltage plateaus at about 2.18 V and 2.06 V (ΔV = 0.12 V), respectively. The specific discharge capacity of the NTP/CNFs half cell recorded at a current rate of 0.05C was around 131 mA h g−1, which corresponded to 98.5% of its theoretical capacity based on the weight of NTP. Because no metal current collector was used for the electrode, the specific capacity based on the total weight of the NTP/CNFs electrode reached an outstanding value of 101 mA h g−1. In the case of the NVP/CNFs half cell, reversible charge and discharge voltage plateaus were observed at around 3.38 V and 3.27 V (ΔV = 0.11 V), respectively. The specific discharge capacities of the NVP/CNFs half cell calculated from the weight of NVP and the total weight of the electrode were about 114 mA h g−1 and 86 mA h g−1, respectively, which corresponded to 97.4% of its theoretical capacity.
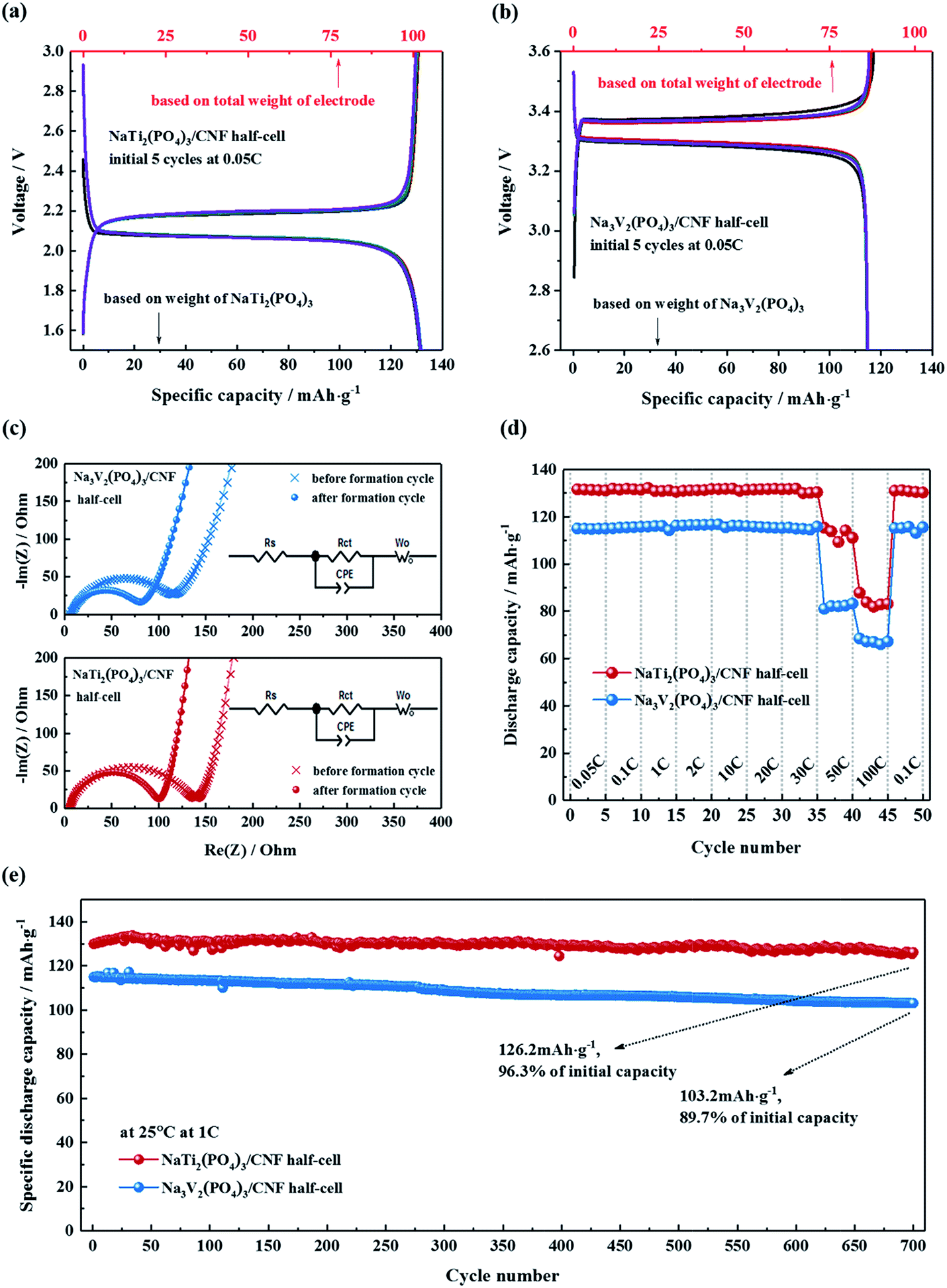 | ||
| Fig. 4 Electrochemical performance of NaTi2(PO4)3/CNFs and Na3V2(PO4)3/CNFs electrodes in half cells. Charge–discharge profiles for formation cycles ((a) and (b)), impedance before and after formation cycles (c), rate capability (d) and cycling performance at 1C (e). All the electrochemical measurements were performed at 25 °C. | ||
The changes in the impedance of the NTP/CNFs and NVP/CNFs half cells before and after the five formation cycles were investigated by EIS in the frequency range from 100 kHz to 10 mHz. As shown in Fig. 4c, all the Nyquist plots for the cells exhibited a semicircle in the high-frequency region, which was ascribed to charge transfer processes of sodium ions through the interface between the surface layer of the particles and the electrolyte. The analyses showed that the charge transfer resistance (Rct) of NTP/CNFs and NVP/CNFs decreased slightly from 145 and 120 Ω before the formation cycles to 104 and 82 Ω after the formation cycles, respectively. The low resistances indicated sufficient electrode/electrolyte contacts and efficient intrinsic electron/ion transport in the self-standing electrodes. The straight lines in the low-frequency region in the Nyquist plots were attributed to the diffusion of sodium ions in the bulk of the electrode material, and the value of the diffusion coefficient (DNa+) for sodium ions can be estimated using the equation DNa+ = 0.5(RT/An2F2σwC)2, where R is the gas constant, T is the temperature, A is the area of the electrode surface (according to the BET results), n is the number of electrons per species reaction, F is the Faraday constant, σw is the Warburg factor, and C is the molar concentration of Na+ ions.47 The calculated values of the diffusion coefficient for sodium (based on the EIS data obtained after the formation cycles) for NTP/CNFs and NVP/CNFs were 9.18 × 10−11 and 6.25 × 10−12 cm2 s−1, respectively, which are slightly higher than previously reported values48–51 and demonstrate the capacity for fast transport of Na+ ions of the prepared electrodes.
The rate capability of the half cells was further examined over subsequent cycles from 0.05C to 100C, as shown in Fig. 4d. With an increase in the current rate, both electrodes exhibited stable capacities in each state. When the current rate was increased to 30C, both electrodes were still able to deliver more than 95% of their theoretical capacities. Even at a current rate of 100C, more than half of the theoretical capacities of both electrodes could still be delivered. When the cycling rate was reduced to 0.1C again, almost 100% of their initial capacities were recovered. These results for high rate capability suggest the advantages of NASICON-type electrodes that are enhanced by CNFs. For automotive applications, a maximum current rate of 7C is required for battery systems. Hence, the NTP/CNFs and NVP/CNFs, with their excellent rate capability, are potential candidates for practical automotive batteries.
The long-term cycling performance of the NTP/CNFs and NVP/CNFs half cells at a current rate of 1C is shown in Fig. 4e. Both electrodes could be cycled for up to 700 cycles at 1C with stable discharge capacities. After 700 cycles, the NTP/CNFs half cell delivered 126.2 mA h g−1 with a capacity retention of 96.3% and the NVP/CNFs half cell delivered 103.2 mA h g−1 with a capacity retention of 89.7%, which illustrated that the prepared electrodes exhibited high chemical and structural stability for prolonged sodiation/desodiation in the relevant voltage ranges. The high capacity based on the total weight of the electrode, high rate capability and long lifespan of the prepared self-standing electrodes can be attributed to the unique morphology of the active particles in/on the CNFs, as well as the highly stable structural framework of NASICON-type materials.9 Specifically, the two steps of mass loading enabled a high energy-to-mass ratio of the electrodes. The uniformly distributed active materials with small particle sizes in the CNFs resulted in a large specific surface area and thus provided better contact between the electrode materials and the electrolyte. The CNTs and carbon layers on the surface of particles of NTP and NVP provided a continuous network that guaranteed electronic conductivity. The NASICON structure formed a stable host framework for the fast and long-term extraction/reinsertion of Na+. These features of the prepared electrodes provided the basis for their superior individual electrochemical properties.
On the basis of the above results, all-phosphate full cells were assembled using NTP/CNFs as the anode, NVP/CNFs as the cathode and 1 M NaPF6 in EC:PC (1:1, vol./vol.) as a liquid electrolyte. The electrochemical compatibility of the electrode pair was examined using CV curves at various scan rates in the voltage range of 0.5–2.0 V, as shown in Fig. S5a.† One well-defined redox couple (Ia and Ia′) was observed in each scan, which can be attributed to the deintercalation/intercalation of two Na ions from/into NVP/CNFs into/from NTP/CNFs. The intensities of the peak currents gradually increased with an increase in the scan rate, with a progressive shift in the redox potentials. Despite the complex nature of the electrode processes when CNFs are present, it is important to determine whether the electrode reaction is dominated by faradaic insertion and extraction of sodium or by capacitive storage of sodium. According to the relationship between the redox current and the scan rate, whereas a linear correlation of the current (I) with the scan rate (v) implies a capacitive process, a proportional correlation between the current (I) and the square root of the scan rate (v1/2) indicates a diffusion-controlled process characteristic of a faradaic insertion and extraction process of sodium.52,53 Both relationships were subsequently plotted in Fig. S5b and c,† whereby it became apparent that the latter mechanism prevailed in the NTP/CNFs||NVP/CNFs electrode pair. In addition, even at a scan rate of 10 mV s−1, the oxidation and reduction polarization at the cut-off voltages was low, which revealed that the uniform distribution of Na+ and electrons in the electrodes benefited from the large specific surface area, small size of the active materials and sufficient electronic conductivity. To further investigate the behavior of CNTs in the NTP/CNFs and NVP/CNFs electrodes during electrochemical processes, in situ Raman studies were carried out on both electrodes in all-phosphate full cells. The cells were cycled with a low current of 0.09 mA (∼0.1C) in the voltage range of 0.5–2.0 V. At the same time, Raman spectra were recorded for one complete sodiation–desodiation process. As shown in Fig. 5, there were negligible changes in the characteristic Raman peaks of CNTs in both electrodes during the initial cycle. These results confirmed that the CNFs in the prepared self-standing electrodes served for electron transport and as a flexible matrix for the electrode materials, rather than as hosts for Na+.
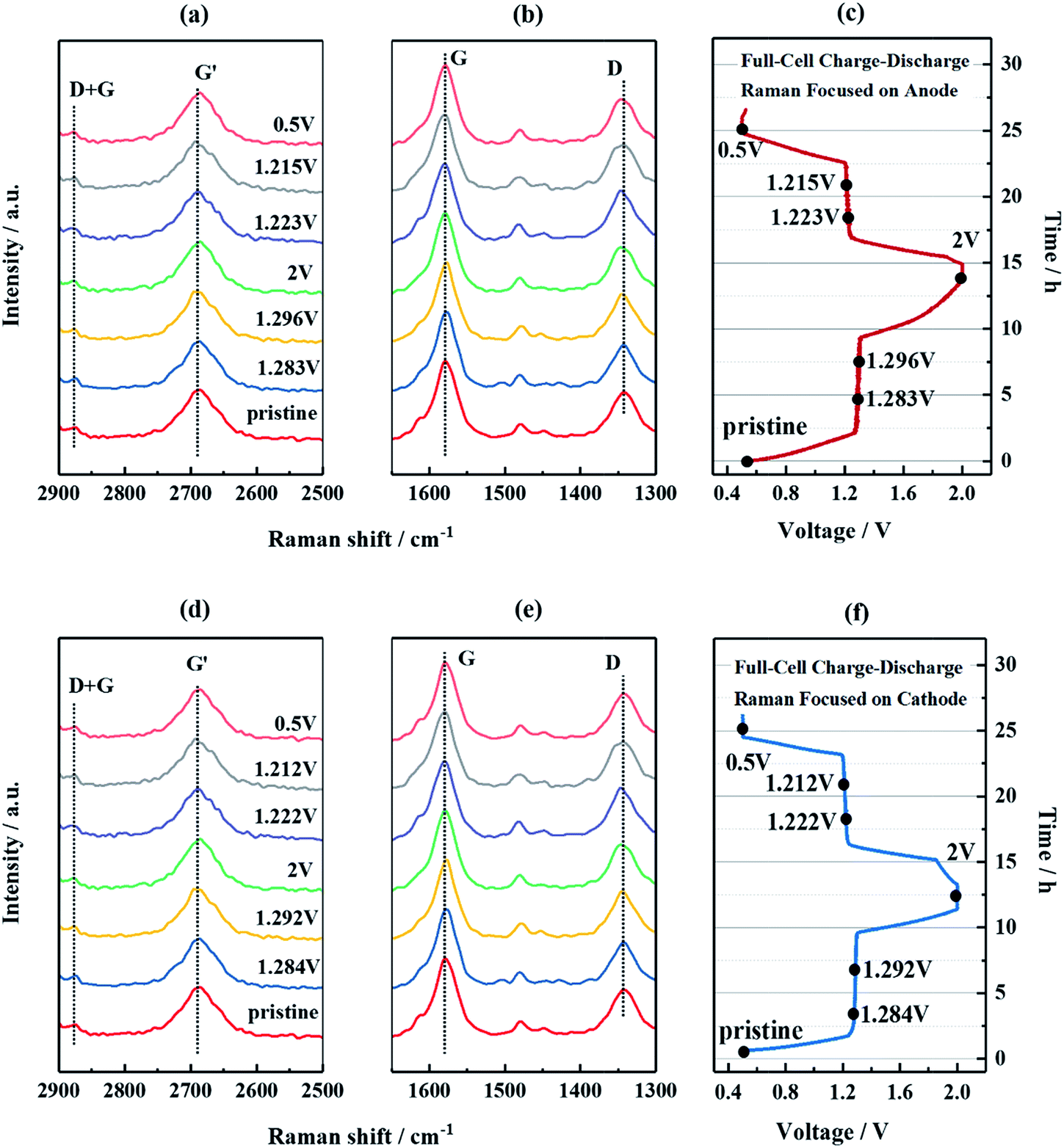 | ||
| Fig. 5 In situ Raman spectra and corresponding time–voltage profiles of the NaTi2(PO4)3/CNFs||Na3V2(PO4)3/CNFs full cell. The Raman spectra were recorded for the NaTi2(PO4)3/CNFs anode ((a)–(c)) and the Na3V2(PO4)3/CNFs cathode ((d)–(f)), respectively. The cells were cycled in the voltage range of 0.5–2.0 V. | ||
The electrochemical performance of the all-phosphate full cells was examined in Swagelok cells, and the results are shown in Fig. 6. The NTP/CNFs and NVP/CNFs electrodes were balanced with a capacity ratio of 1:1.2, and thus the specific capacity of the cells could be calculated from the weight of the active materials in the anode. The battery was initially cycled at a low current rate of 0.05C for formation of the cell. The charge–discharge profiles for the initial three cycles, as shown in Fig. 6a, exhibited a potential plateau around 1.3 V with a discharge capacity of 126 mA h g−1. The coulombic efficiency was 93.6% in the first cycle and increased to around 99.5% in the subsequent cycles. The low coulombic efficiency in the first cycle was due to the formation of SEI and activation of the battery.54–56 Benefiting from the high mass loadings of the electrodes, the discharge capacities based on the total weight of the anode and both electrodes were 100 and 42 mA h g−1, respectively, which demonstrated a promising application of the electrodes for flexible SIBs with a high energy density. Furthermore, after the formation of the batteries, as shown in Fig. 6b, the charge transfer resistance of the cell displayed only a slight decrease, which was consistent with the behavior of the half cells, which proved the structural and electrical integrity of the phosphate electrodes and their interfaces with the electrolyte.
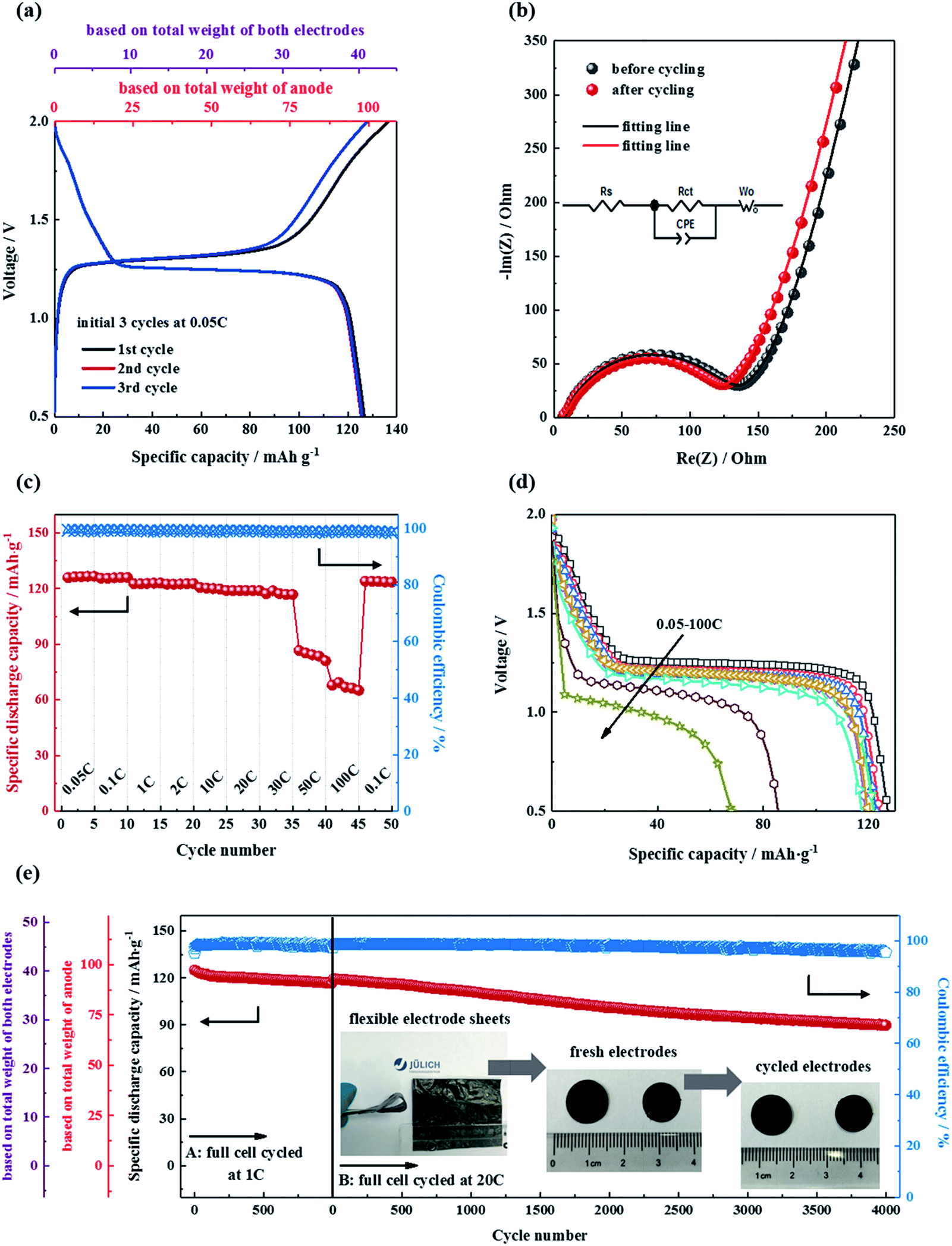 | ||
| Fig. 6 Electrochemical performance of NaTi2(PO4)3/CNFs||Na3V2(PO4)3/CNFs full cells. Charge–discharge profiles for formation cycles (a), Nyquist plots of impedance before and after formation cycles (b), rate capability (c) and corresponding discharge curves (d) and cycling stability of two full cells at 1C and 20C, respectively (e). All the electrochemical measurements were performed at 25 °C. | ||
The all-phosphate battery was charged/discharged at various current rates from 0.05C to 100C for five cycles in each state (Fig. 6c). All cycles exhibited a coulombic efficiency greater than 99%. Stabilized specific discharge capacities of around 120 mA h g−1 were delivered up to 30C, which corresponded to more than 90% of the theoretical capacity. With a further increase in the current rate to 50 and 100C, the specific discharge capacities decreased to 85 and 68 mA h g−1 correspondingly. The initial specific discharge capacity of 122 mA h g−1 was recovered when the rate was returned to 0.1C. The discharge voltage profiles of the battery at different current rates are shown in Fig. 6d. Voltage plateaus are clearly revealed in the discharge process regardless of variations in the current rate, which indicates that the redox reactions of titanium and vanadium took place during the cycling process at all current rates. In addition, the slight decrease in the discharge plateau at high current rates can be attributed to ohmic and electrochemical polarization of the cell components, as well as kinetic limitations due to ion diffusion within the electrolyte and the electrode/electrolyte interface.57,58 Overall, the insignificant polarization at high current rates suggests fast transport of ions and electrons in the electrodes. The long-term cycling stability of the full cell was investigated at current rates of 1C and 20C, respectively, and the results are shown in Fig. 6e. At a moderate current rate (1C), the NTP/CNFs||NVP/CNFs cell exhibited a capacity retention of 91% after 1000 cycles. At a high current rate of 20C, the cycling performance of the battery still maintained a reversible capacity of greater than 90 mA h g−1, which corresponded to a capacity retention of 74.5%, with high coulombic efficiency after cycling 4000 times. The superior rate capability and cycling stability of the all-phosphate battery were in accordance with the results obtained from the half cells, as they benefited from the novel morphology and stable structure of the electrodes. On comparing the NTP/CNFs and NVP/CNFs with previously reported flexible electrode materials for SIBs, as shown in Table S1,† the electrodes prepared in this study were very competitive owing to their significantly high mass loading and outstanding electrochemical performance.
Although the capacity loss in the full cell during cycling was not severe, the capacity fade hints at irreversible changes in the electrodes, which are considered to originate from chemical and structural decomposition and/or the continuous growth of a solid electrolyte interphase (SEI) layer. To understand the mechanism of aging, the full cell after long-term cycling at 20C was disassembled in a glovebox and post-tests were carried out on the electrodes. As shown in Fig. 6e, no damage to the electrodes was observable after long-term cycling. Furthermore, the crystallinity and microstructure of the electrodes were investigated by XRD and TEM, respectively. As shown in Fig. S6,† the XRD diffraction patterns of the cycled NTP/CNFs electrode still indicated a NASICON NaTi2(PO4)3 phase, whereas the NVP/CNFs electrode contained Na3V2(PO4)3 and desodiated NaxV2(PO4)3 phases (x = 1 or 2), which was probably due to a delay in the phase transition of the cathode. Because the NTP/CNFs electrode was the capacity-limiting electrode in the battery and had a higher DNa+ than the NVP/CNFs electrode, when XRD patterns were recorded immediately after long-term fast cycling at a constant current rate of 20C, the phase transition of NVP in the bulk NVP/CNFs may still have been incomplete owing to a difference in concentration polarization. In addition, the desodiated NaxV2(PO4)3 phases indicated that at least part of the Na from the NVP/CNFs was trapped in the SEI layer after long-term cycling. Nevertheless, no crystalline species due to decomposition could be observed in the XRD patterns of either electrode. The bulk microstructures of the electrodes were the same before and after cycling, as confirmed by TEM images (Fig. S7†). The results of post-test measurements indicate the high reversibility and high structural and morphological stability of the electrodes during cycling.
Because ssNMR spectroscopy is a powerful tool for monitoring structural changes and the evolution of species in battery materials,59 further investigations into possible changes in NTP/CNFs and NVP/CNFs during cycling were performed via in operando23Na ssNMR measurements with a freshly assembled battery after two formation cycles at 0.05C. Before the in operando23Na ssNMR measurements, the three components of the full cell were studied individually, and their NMR spectra are shown in Fig. S8.† The shifts and line shapes of the resonances of NTP/CNFs and NVP/CNFs were similar, and both resonances were present around 0 ppm with a broad linewidth in comparison with the narrow resonance of the electrolyte, which appeared at −13 ppm.
As shown in Fig. 7a, the specific discharge capacities measured with the in operando NMR battery were highly comparable to the results obtained in the Swagelok cells. The initial discharge capacity was 122 mA h g−1 and continuously declined to 93 mA h g−1, which corresponded to a capacity loss of 23.7%, in the last cycle after charging and discharging 1000 times at a high current rate of 10C. On comparing the contour plot for the in operando NMR spectra recorded in the initial cycle with that from the 1002nd cycle, as shown in Fig. 7b and c, the intensity between 10 and −40 ppm increased dramatically whereas that in the other ranges stayed more or less the same, which implies that a resonance appeared/increased during cycling. Normally, the NMR resonance of SEI layer in Na-ion batteries is located between 0 and −30 ppm.60 From the XRD and TEM results, we can conclude that both electrodes maintained their structure and morphology after long-term cycling. Hence, the difference in the NMR spectra between the 1st and 1002nd cycles can be assigned to the formation of SEI layer.
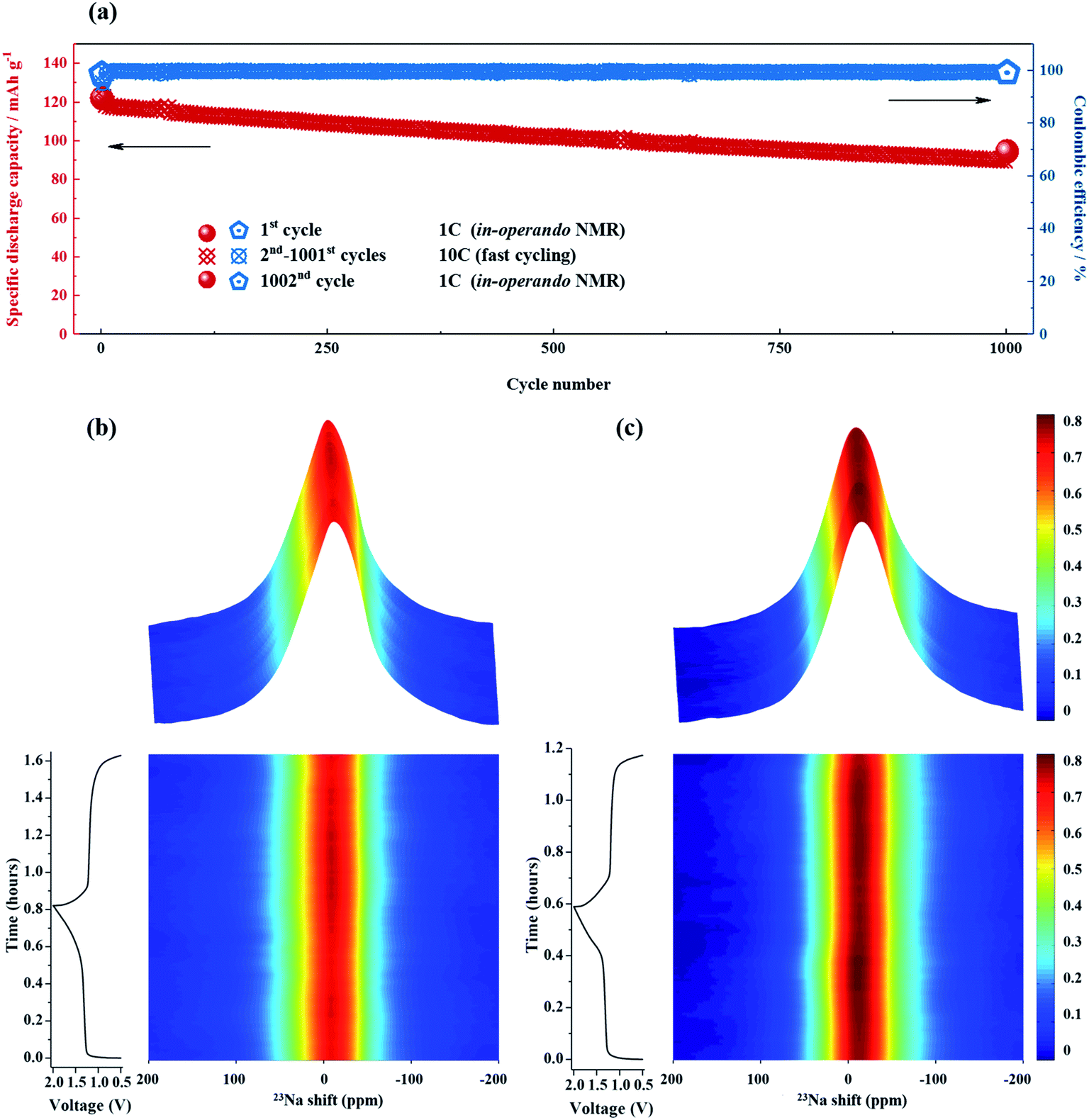 | ||
| Fig. 7 (a) Specific discharge capacity and coulombic efficiency of NaTi2(PO4)3/CNFs||Na3V2(PO4)3/CNFs full cell in an in operando NMR battery. The current rates were ∼1C in the initial and last cycles and ∼10C in the intervening 1000 cycles. (b) and (c) Results of in operando23Na NMR spectroscopy and corresponding voltage–time curves for the initial and last cycles. The NMR and electrochemical tests were performed at 30 °C. | ||
The above results showed the excellent rate capability and long lifespan of the self-standing electrodes. The two aspects of flexibility and high volumetric energy density were investigated regarding practical applications of the prepared electrodes and all-phosphate SIBs in the following section. For the flexibility tests, a prototype of a pouch cell consisting of a NTP/CNFs anode and a NVP/CNFs cathode was prepared (Fig. 8a and b). The sizes of the electrodes were 4.5 cm × 7 cm for the anode and 5 cm × 9 cm for the cathode, which had the same thickness of 130 μm. The theoretical capacity of the cell was about 38 mA h. Al and Cu tapes, which both had a small size of 0.8 cm × 14 cm, were used as external contact tabs to guarantee a flow of current to the cathode and anode, respectively. The electrolyte was slowly injected into a glass fiber separator between the electrodes to remove gas in the pouch bag. As can be seen in the photographs in Fig. 8, the flexibility of the electrodes was inherited by the pouch cell. The cell exhibited a lower specific capacity, shorter charge–discharge voltage plateaus and higher polarization in comparison with the results obtained from Swagelok cells with the same electrodes with smaller sizes. The total capacities of the cell were 26.9, 26.4 and 24.7 mA h at 0.1, 0.2 and 0.5C, respectively, which corresponded to 65–70% of the theoretical capacity. Although the full ability of the electrode pair in terms of its electrochemical performance cannot be efficiently demonstrated in such a prototype battery owing to the limited locations for the tabs and the lack of external pressure on the large-format flexible pouch cell,61 it is clear that the electrodes are promising for applications in flexible electronic devices.
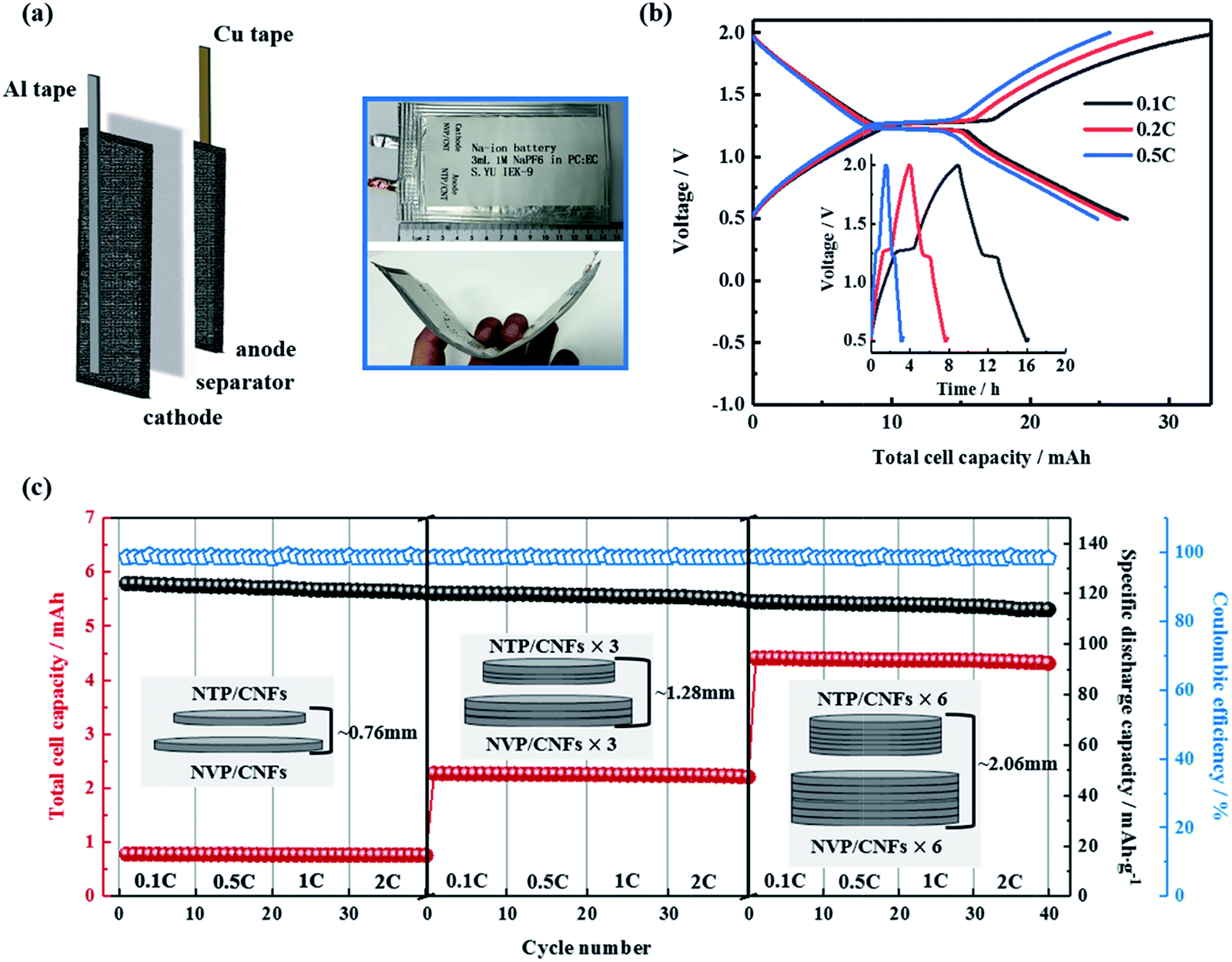 | ||
| Fig. 8 (a) Schematic diagram and photographs of NaTi2(PO4)3/CNFs||Na3V2(PO4)3/CNFs prototype flexible pouch cell. (b) Charge–discharge curves and corresponding time–voltage profiles of the pouch cell. (c) Rate capability of batteries consisting of 1, 3 and 6 electrode pairs. The stacked electrodes were prepared by simply stacking three or six of the as-prepared electrodes in Swagelok-type battery cases. The NaTi2(PO4)3/CNFs and Na3V2(PO4)3/CNFs were immersed in a liquid electrolyte for 24 h before use to fully infiltrate the bulk and surface of the electrodes. | ||
For the fabrication of electrodes with a high volumetric energy density, the usual method involves choosing active materials with a large particle size and high tap density to form thick electrodes,62–64 which all have issues that hinder the rate performance and cycling life of a battery. Using self-standing electrodes with high mass loadings without a metal current collector, an easy approach comprises simply stacking multiple layers of electrodes in one cell. Hence, the volumetric energy density can be greatly increased in a simple way if the full capacity of each electrode layer is delivered. With this aim, Swagelok cells with one, three and six pairs of electrodes were prepared and measured. Fig. 8c shows schematic diagrams and the rate capabilities of these cells. The specific discharge capacities recorded for one, three and six electrodes in a stack were almost constant even at a current rate of 2C. The total capacity of the cells increased severalfold from 0.78 mA h for a single electrode pair to 2.24 and 4.37 mA h for three and six pairs, which corresponded to 93.2%, 89.2% and 87.0% of the theoretical capacities of the cells, respectively. The volumetric capacities of the cells were about 12.2, 15.1 and 23.2 mA h cm−3, respectively. Therefore, it can be concluded that the conductivity of the self-standing electrodes is sufficient without a metal current collector and the energy density of SIBs can be increased by simply stacking multiple electrodes. Finally, on the basis of the results of feasibility tests, the self-standing NTP/CNFs and NVP/CNFs electrodes with high mass loadings and the corresponding all-phosphate SIB are proven to be promising for practical applications.
Moreover, Fig. S9† shows Ragone plots of the energy and power densities of the all-phosphate batteries (half cells and full cell), together with conventional energy storage devices. The cells presented in this work exhibited a sufficient gravimetric energy density and high gravimetric power density on a device level (Fig. S9a†) to meet the requirements for batteries used in hybrid electric vehicles. The lower gravimetric specific energy density of SIBs in comparison with Li-ion batteries is due to the high atomic mass of Na (vs. Li) and the low voltage of the battery. In fact, as shown in Fig. S9b,† the energy density of the phosphate cells based on the device volume was comparable to that of Li-ion batteries. The volumetric power density of the prepared batteries was about two orders of magnitude higher than that of conventional Li-ion batteries and one order of magnitude higher than that of a high-power commercial battery. In addition, the NTP/CNFs and NVP/CNFs electrodes with high mass loadings, which were directly formed by crystallization of the active materials, are easily scalable and relatively cheap in terms of energy per gram, because the additives and traditional processes for the preparation of conventional electrodes are not needed.
Conclusions
In summary, an all-phosphate SIB with self-standing electrodes of NTP/CNFs and NVP/CNFs has been prepared and achieved high electrochemical performance. These electrodes possessed high mass loading and could be directly used to fabricate SIBs without using a metal current collector, an inactive binder and additional conductive carbon, as in the conventional process of electrode manufacture. The carbon nanotubes in the electrodes served as a 3D network for electron conduction and as a flexible matrix to accommodate the active materials. The NTP/CNFs anode and NVP/CNFs cathode were characterized in half cells, in which their high rate capability was estimated. Even at a rate of 30C, more than 97% of their theoretical capacities were delivered by both NTP/CNFs and NVP/CNFs. Moreover, the all-phosphate full cell exhibited a discharge capacity of 66 mA h g−1 at an extremely high rate of 100C. Furthermore, the full cell displayed a capacity retention of 92% at 1C over 1000 cycles, followed by outstanding high rate cycling performance at 20C, with a reversible capacity of about 90 mA h g−1 after 4000 cycles. The slight capacity fade over long-term cycling was due to the growth of SEI layer, which was confirmed by post-test XRD, TEM and in operando NMR measurements. Besides, good electrochemical performance was demonstrated by the NTP/CNFs and NVP/CNFs electrodes in prototype flexible pouch cells and Swagelok batteries with multiple electrodes. The self-standing electrodes displayed promising electrochemical properties in terms of a high specific capacity, high rate capability and long cycling life and revealed great potential for practical applications in a flexible SIB with high energy and power densities. Moreover, the simple and scalable synthesis strategy developed in this study can be easily extended to prepare other self-standing electrodes for energy storage devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the projects of “Materials and Components to Meet High Energy Density Batteries II” of the funding program “Excellent battery” from the Bundesministerium für Bildung und Forschung (BMBF) within the framework of the program “Materials Innovations for Industry and Society” (project no. 03XP0084C). The carbon nanotube fabrics were supplied by Tortech Nano-Fibers Ltd. (Israel) free of charge, the authors are grateful to this. Dr Shicheng Yu is grateful to Dr Roland Schierholz and Daniel Kröger, who helped in the SEM and TEM training. Dr Shicheng Yu is grateful to Dr Mengchu Tao for her support in the past and in the future.Notes and references
- S. Bauer, Nat. Mater., 2013, 12, 871–872 CrossRef PubMed
.
- Z. F. Liu, S. Fang, F. A. Moura, J. N. Ding, N. Jiang, J. Di, M. Zhang, X. Lepró, D. S. Galvão, C. S. Haines, N. Y. Yuan, S. G. Yin, D. W. Lee, R. Wang, H. Y. Wang, W. Lv, C. Dong, R. C. Zhang, M. J. Chen, Q. Yin, Y. T. Chong, R. Zhang, X. Wang, M. D. Lima, R. Ovalle–Robles, D. Qian, H. Lu and R. H. Baughman, Science, 2015, 349, 400–404 CrossRef PubMed
- C. J. Zhang, S. H. Park, O. Ronan, A. Harvey, A. Seral–Ascaso, Z. Lin, N. McEvoy, C. S. Boland, N. C. Berner, G. S. Duesberg, P. Rozier, J. N. Coleman and V. Nicolosi, Small, 2017, 13, 1–11 Search PubMed
- T. Hoshide, Y. Zheng, J. Hou, Z. Wang, Q. Li, Z. Zhao, R. Ma, T. Sasaki and F. Geng, Nano Lett., 2017, 17, 3543–3549 CrossRef PubMed
- S. Chen, C. Wu, L. Shen, C. Zhu, Y. Huang, K. Xi, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1–21 Search PubMed
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 RSC
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 1–10 Search PubMed
- D. Yu, Q. Pang, Y. Gao, Y. Wei, C. Wang, G. Chen and F. Du, Energy Storage Mater., 2018, 11, 1–7 CrossRef
- S. Chen, L. Shen, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1–21 Search PubMed
- C. Zhu, K. Song, P. A. Van Aken, J. Maier and Y. Yu, Nano Lett., 2014, 14, 2175–2180 CrossRef PubMed
- G. Yang, H. Song, M. Wu and C. Wang, J. Mater. Chem. A, 2015, 3, 18718–18726 RSC
- Q. Liu, D. Wang, X. Yang, N. Chen, C. Wang, X. Bie, Y. Wei, G. Chen and F. Du, J. Mater. Chem. A, 2015, 3, 21478–21485 RSC
- F. Lalère, V. Seznec, M. Courty, R. David, J. N. Chotard and C. Masquelier, J. Mater. Chem. A, 2015, 3, 16198–16205 RSC
- D. Wang, X. Bie, Q. Fu, D. Dixon, N. Bramnik, Y. S. Hu, F. Fauth, Y. Wei, H. Ehrenberg, G. Chen and F. Du, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed
- W. Zhang, Y. Liu, C. Chen, Z. Li, Y. Huang and X. Hu, Small, 2015, 11, 3822–3829 CrossRef PubMed
- Y. Sun and J. A. Rogers, Adv. Mater., 2007, 19, 1897–1916 CrossRef
- S. Yue, H. Tong, L. Lu, W. Tang, W. Bai, F. Jin, Q. Han, J. He, J. Liu and X. Zhang, J. Mater. Chem. A, 2017, 5, 689–698 RSC
- X. Rui, W. Sun, C. Wu, Y. Yu and Q. Yan, Adv. Mater., 2015, 27, 6670–6676 CrossRef PubMed
- S. Yehezkel, M. Auinat, N. Sezin, D. Starosvetsky and Y. Ein–Eli, J. Power Sources, 2016, 312, 109–115 CrossRef
- D. Guo, J. Qin, Z. Yin, J. Bai, Y. K. Sun and M. Cao, Nano Energy, 2018, 45, 136–147 CrossRef
- W. Wang, Q. Xu, H. Liu, Y. Wang and Y. Xia, J. Mater. Chem. A, 2017, 5, 8440–8450 RSC
- L. David, R. Bhandavat and G. Singh, ACS Nano, 2014, 8, 1759–1770 CrossRef PubMed
- Y. Fang, L. Xiao, X. Ai, Y. Cao and H. Yang, Adv. Mater., 2015, 27, 5895–5900 CrossRef PubMed
- M. Kim, C. Lee and J. Jang, Adv. Funct. Mater., 2014, 24, 2489–2499 CrossRef
- Z. Zhang, F. Xiao, L. Qian, J. Xiao, S. Wang and Y. Liu, Adv. Energy Mater., 2014, 4, 1400064 CrossRef
- M. Jing, H. Zhai, Z. Pi, J. Li, L. Chen, X. Shen, X. Xi and K. Xiao, Russ. J. Appl. Chem., 2016, 89, 1183–1188 CrossRef
- S. Li, G. Liu, J. Liu, Y. Lu, Q. Yang, L. Y. Yang, H. R. Yang, S. Liu, M. Lei and M. Han, J. Mater. Chem. A, 2016, 4, 6426–6432 RSC
- S. A. Kayser, A. Mester, A. Mertens, P. Jakes, R. A. Eichel and J. Granwehr, Phys. Chem. Chem. Phys., 2018, 20, 13765–13776 RSC
- C. Sole, N. E. Drewett and L. J. Hardwick, Faraday Discuss., 2014, 172, 223–237 RSC
- L. J. Hardwick, M. Hahn, P. Ruch, M. Holzapfel, W. Scheifele, H. Buqa, F. Krumeich, P. Novák and R. Kötz, Electrochim. Acta, 2006, 52, 675–680 CrossRef
- Y. Liu, X. Fang, M. Ge, J. Rong, C. Shen, A. Zhang, H. A. Enaya and C. Zhou, Nano Energy, 2015, 16, 399–407 CrossRef
- W. Ren, W. Zhou, H. Zhang and C. Cheng, ACS Appl. Mater. Interfaces, 2017, 9, 487–495 CrossRef PubMed
- W. Ren, H. Zhang, C. Guan and C. Cheng, Adv. Funct. Mater., 2017, 27, 1702116 CrossRef
- S. Yu, A. Mertens, R. Schierholz, X. Gao, Ö. Aslanbas, J. Mertens, H. Kungl, H. Tempel and R.-A. Eichel, J. Electrochem. Soc., 2017, 164, A370–A379 CrossRef
- S. Yu, A. Mertens, H. Kungl, R. Schierholz, H. Tempel and R.-A. Eichel, Electrochim. Acta, 2017, 232, 310–322 CrossRef
- C. Wu, P. Kopold, Y. L. Ding, P. A. Van Aken, J. Maier and Y. Yu, ACS Nano, 2015, 9, 6610–6618 CrossRef PubMed
- D. Wang, Q. Liu, C. Chen, M. Li, X. Meng, X. Bie, Y. Wei, Y. Huang, F. Du, C. Wang and G. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 2238–2246 CrossRef PubMed
- B. Zhao, Q. Wang, S. Zhang and C. Deng, J. Mater. Chem. A, 2015, 3, 12089–12096 RSC
- H. Geng, J. Yang, H. Yu, C. Li and X. Dong, Mater. Chem. Front., 2017, 1, 1435–1440 RSC
- J. Mao, C. Luo, T. Gao, X. Fan and C. Wang, J. Mater. Chem. A, 2015, 3, 10378–10385 RSC
- S. Tao, X. Wang, P. Cui, Y. Wang, Y. A. Haleem, S. Wei, W. Huang, L. Song and W. Chu, RSC Adv., 2016, 6, 43591–43597 RSC
- S. Yu, H. Tempel, R. Schierholz, Ö. Aslanbas, X. Gao, J. Mertens, L. G. J. de Haart, H. Kungl and R.-A. Eichel, ChemElectroChem, 2016, 3, 1157–1169 CrossRef
- Y. Liu, A. Zhang, C. Shen, Q. Liu, X. Cao, Y. Ma, L. Chen, C. Lau, T. C. Chen, F. Wei and C. Zhou, ACS Nano, 2017, 11, 5530–5537 CrossRef PubMed
- J. Liang, K. Fan, Z. Wei, X. Gao, W. Song and J. Ma, Mater. Res. Bull., 2018, 99, 343–348 CrossRef
- W. Shen, H. Li, Z. Guo, C. Wang, Z. Li, Q. Xu, H. Liu, Y. Wang and Y. Xia, ACS Appl. Mater. Interfaces, 2016, 8, 15341–15351 CrossRef PubMed
- C. Xu, Y. Xu, C. Tang, Q. Wei, J. Meng, L. Huang, L. Zhou, G. Zhang, L. He and L. Mai, Nano Energy, 2016, 28, 224–231 CrossRef
- K. Du, H. Guo, G. Hu, Z. Peng and Y. Cao, J. Power Sources, 2013, 223, 284–288 CrossRef
- Z. Huang, L. Liu, L. Yi, W. Xiao, M. Li, Q. Zhou, G. Guo, X. Chen, H. Shu, X. Yang and X. Wang, J. Power Sources, 2016, 325, 474–481 CrossRef
- J. Liu, K. Tang, K. Song, P. A. Van Aken, Y. Yu and J. Maier, Nanoscale, 2014, 6, 5081–5086 RSC
- N. Böckenfeld and A. Balducci, J. Solid State Electrochem., 2014, 18, 959–964 CrossRef
- S. Li, Y. Dong, L. Xu, X. Xu, L. He and L. Mai, Adv. Mater., 2014, 26, 3545–3553 CrossRef PubMed
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P. L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef PubMed
- J. Liu, M. Chen, L. Zhang, J. Jiang, J. Yan, Y. Huang, J. Lin, H. J. Fan and Z. X. Shen, Nano Lett., 2014, 14, 7180–7187 CrossRef PubMed
- M. Dahbi, T. Nakano, N. Yabuuchi, S. Fujimura, K. Chihara, K. Kubota, J. Y. Son, Y. T. Cui, H. Oji and S. Komaba, ChemElectroChem, 2016, 3, 1856–1867 CrossRef
- J. Sun, H. W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980–985.13 CrossRef
- L. Zhao, J. Zhao, Y. S. Hu, H. Li, Z. Zhou, M. Armand and L. Chen, Adv. Energy Mater., 2012, 2, 962–965 CrossRef
- S. Yu, A. Mertens, H. Tempel, R. Schierholz, H. Kungl and R. –A. Eichel, ACS Appl. Mater. Interfaces, 2018, 10, 22264–22277 CrossRef PubMed
-
A. Bard and L. R. Faulkner, Electrochemical Methods, Wiley press, New York, 2nd edn, 2001, pp. 156–225 Search PubMed
- O. Pecher, J. Carretero–Gonzalez, K. J. Griffith and C. P. Grey, Chem. Mater., 2017, 29, 213–242 CrossRef
- J. M. Stratford, M. Mayo, P. K. Allan, O. Pecher, O. J. Borkiewicz, K. M. Wiaderek, K. W. Chapman, C. J. Pickard, A. J. Morris and C. P. Grey, J. Am. Chem. Soc., 2017, 139, 7273–7286 CrossRef PubMed
- A. Samba, N. Omar, H. Gualous, O. Capron, P. Van Den Bossche and J. Van Mierlo, Electrochim. Acta, 2014, 147, 319–329 CrossRef
- H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano Lett., 2011, 11, 2644–2647 CrossRef PubMed
- H. Zheng, J. Li, X. Song, G. Liu and V. S. Battaglia, Electrochim. Acta, 2012, 71, 258–265 CrossRef
- H. Zhao, Y. Wei, R. Qiao, C. Zhu, Z. Zheng, M. Ling, Z. Jia, Y. Bai, Y. Fu, J. Lei, X. Song, V. S. Battaglia, W. Yang, P. B. Messersmith and G. Liu, Nano Lett., 2015, 15, 7927–7932 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta07313a |
| This journal is © The Royal Society of Chemistry 2018 |