
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A bright outlook on organic photoelectrochemical cells for water splitting
Ludmilla
Steier
 * and
Sarah
Holliday
* and
Sarah
Holliday
Department of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: l.steier@imperial.ac.uk
First published on 8th October 2018
Abstract
Photoelectrochemcial (PEC) water splitting is increasingly attracting attention as a means to generate clean, renewable hydrogen fuel from solar energy. This Highlight article covers the key advances that have been made over recent years in organic photoelectrochemical (OPEC) cell research, and identifies a pathway forwards combining state-of-the-art materials and device engineering from both the organic photovoltaics and inorganic PEC water splitting communities. We discuss the advantages of using buried junction device architectures in OPEC photoelectrodes and identify the need for new materials for OPEC water splitting cells in order to improve their efficiency and operating stability. We present an overview on promising new absorbers and device architectures employed in the parallel field of organic photovoltaics with a critical view on requirements for OPEC water splitting. We also elaborate on progress made with organic multijunction cells that we see as promising photocathode designs for voltage-unassisted water splitting. Finally, we see the urgent need for benchmarking rules, in terms of performance and stability parameters, as this emerging field of OPEC water splitting progresses.
 Ludmilla Steier | Ludmilla Steier obtained her PhD in Chemistry and Chemical Engineering at École Polytechnique Fédérale de Lausanne (EPFL) in 2016 working in the group of Michael Grätzel on thin film materials for photovoltaics and solar fuel production. She then joined the group of James Durrant at Imperial College London where she obtained a Marie Skłodowska Curie Fellowship in 2017. With a broad expertise in fabrication and characterisation of materials, Ludmilla is interested in understanding device physics, materials properties and especially charge transfer across interfaces in order to achieve more efficient and stable devices for solar energy conversion and storage. |
 Sarah Holliday | Sarah Holliday studied Chemistry at the University of Edinburgh before joining the group of Iain McCulloch at Imperial College London in 2011. Here she carried out her PhD research in the design and synthesis of non-fullerene acceptors for organic photovoltaics. She then worked with Christine Luscombe at the University of Washington as a postdoctoral researcher studying the photooxidative stability of organic photovoltaics, as well as developing new materials to improve the performance of CZTS based solar cells. She is particularly interested in the scale-up of low-cost, printed solar energy technologies for real-world applications, especially in the context of energy access globally. |
Introduction
In order to reach commercial application, photoelectrochemical (PEC) water splitting cells must produce hydrogen with high efficiency and long operating stability at low cost. Achieving these three requirements with one photoabsorbing material remains challenging, and there is intensive research in various aspects of PEC systems using traditional inorganic materials such as compound semiconductors (III–V, Si, etc.),1 transition metal oxide (BiVO4, Fe2O3, TiO2, Cu2O) or chalcogenides (CZTS, CIGS).2 More recently, organic semiconductors have been attracting interest within the PEC field due to their unique opportunities to carefully tune the visible light absorption and frontier molecular orbital energies, as well as morphological properties. Furthermore, the compatibility of organic semiconductors with low-cost, large-area printing routes offers great potential for these materials to meet the requirements for efficient, stable and low-cost PEC devices.As the field of organic photoelectrochemical (OPEC) cells is relatively young but growing rapidly, there is an opportunity now to highlight key works in the evolution of OPEC water splitting cells and focus these towards the most promising directions for future research. With a clear vision of the requirements for OPEC cells and knowledge in PEC water splitting with inorganic semiconductors, this Highlight review seeks to advocate for the concept of buried bulk heterojunctions (BHJ) as the most advantageous and promising route towards efficient OPEC water splitting cells. With this conclusion, we elaborate on some of the most recent trends and achievements in research on buried BHJs in organic photovoltaics (OPV) and suggest how these advances can directly contribute to improved materials and electrode design in OPEC photocathodes. Furthermore, we suggest key benchmarking rules in the OPEC water splitting community in order to create criteria of comparison between different materials and cell designs.
Thermodynamic requirements for water splitting
The minimum free energy needed for splitting water into hydrogen and oxygen is ΔG = +237.18 kJ mol−1 at 25 °C, which is equal to a required voltage of 1.23 V (=ΔG/nF for n = 2 electrons with F being the Faraday constant).| ΔG = ΔHf − TΔS = 286.03 kJ mol−1 − 0.163 kJ mol−1 K−1T[K] |
However, in practice, this voltage is not sufficient to split water. When overpotential losses at the anode (ηa), cathode (ηc) and in solution due to low ionic conductivity (ηsol) are taken into account, it becomes clear that the final applied voltage must be substantially higher than 1.23 V. The overpotentials at the anode and cathode are highly dependent on the catalyst used and can easily increase the voltage required to drive the water splitting reaction to 1.5–1.8 V, even with the best catalysts used. Hence, it is important to focus on PEC cell designs that can provide photovoltages to sustain standalone (unbiased) water splitting.
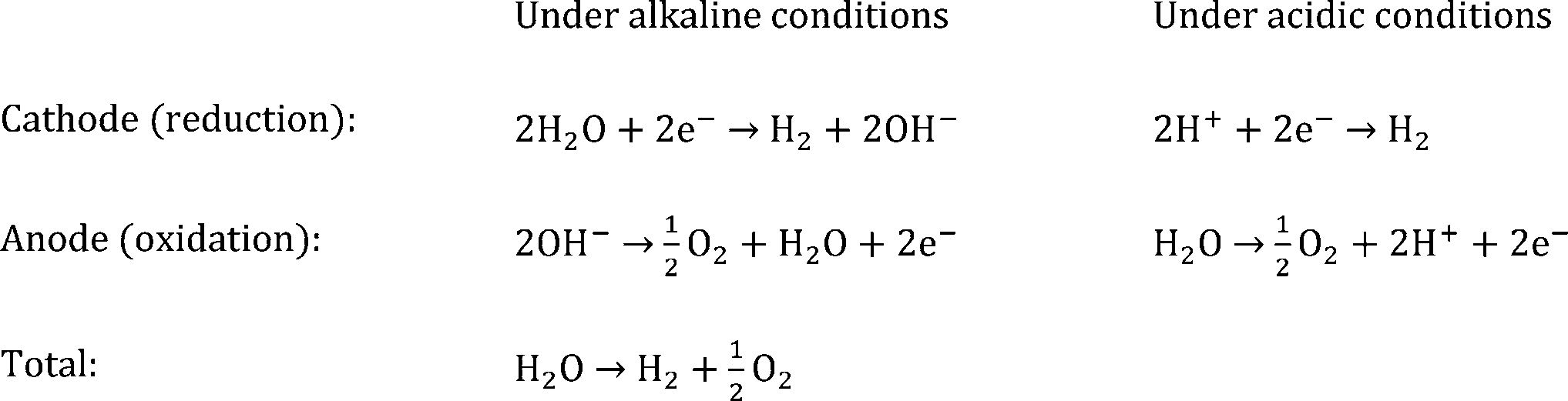
What can be concluded from PEC water splitting with traditional inorganic semiconductors?
Since the research field of OPEC water splitting is relatively young and unexplored, knowledge transfer from research in PEC water splitting with traditional inorganic photoelectrode materials can prove useful. In this paragraph we identify conceptual challenges in inorganic PEC systems and see great opportunities for organic semiconductors to succeed in overcoming those hurdles in OPEC water splitting cells.The research directions of the inorganic PEC water splitting community can conceptually be distinguished by the two types of junctions used: (i) semiconductor/electrolyte solution junctions and (ii) buried junctions wherein the absorber is sandwiched between charge selective contacts and coated with a suitable catalyst. Photoelectrodes based on earth-abundant materials such as BiVO4, hematite or TiO2 make use of the semiconductor/electrolyte junction to drive charge separation and simultaneously use the photoabsorber as the water oxidation catalyst. With the voltage requirement in mind, a big part of the inorganic PEC community is working towards a tandem photocathode-photoanode design shown in Fig. 1a.3 At the photoanode, the n-type semiconductor has to provide enough driving force to spatially separate the charges created upon illumination and efficiently drive the catalysis so that holes are extracted to solution and electrons to the contact before they have time to recombine. However, water oxidation, which requires the accumulation of 3 holes,4 is a very slow reaction (ms to s), while recombination timescales are on the order of ps to ms.5 When the holes at the surface are consumed before they can be extracted, the semiconductor cannot build up the necessary photovoltage, VPh1 to drive the reaction and higher internal electric fields will be required to minimise recombination. A similar challenge is faced by the photocathode though the rate of reaction is faster for hydrogen evolution than for water oxidation. Indeed, photovoltages in these photoelectrodes are significantly lower than the Shockley–Queisser limit (Fig. 1b).6 Commonly used strategies to increase the build-up of photovoltage in the photoabsorber include the use of (i) co-catalysts to improve or enable photocatalysis to occur,7,8 (ii) surface state passivation overlayers9–12 or (iii) capacitive charge storage overlayers.13,14
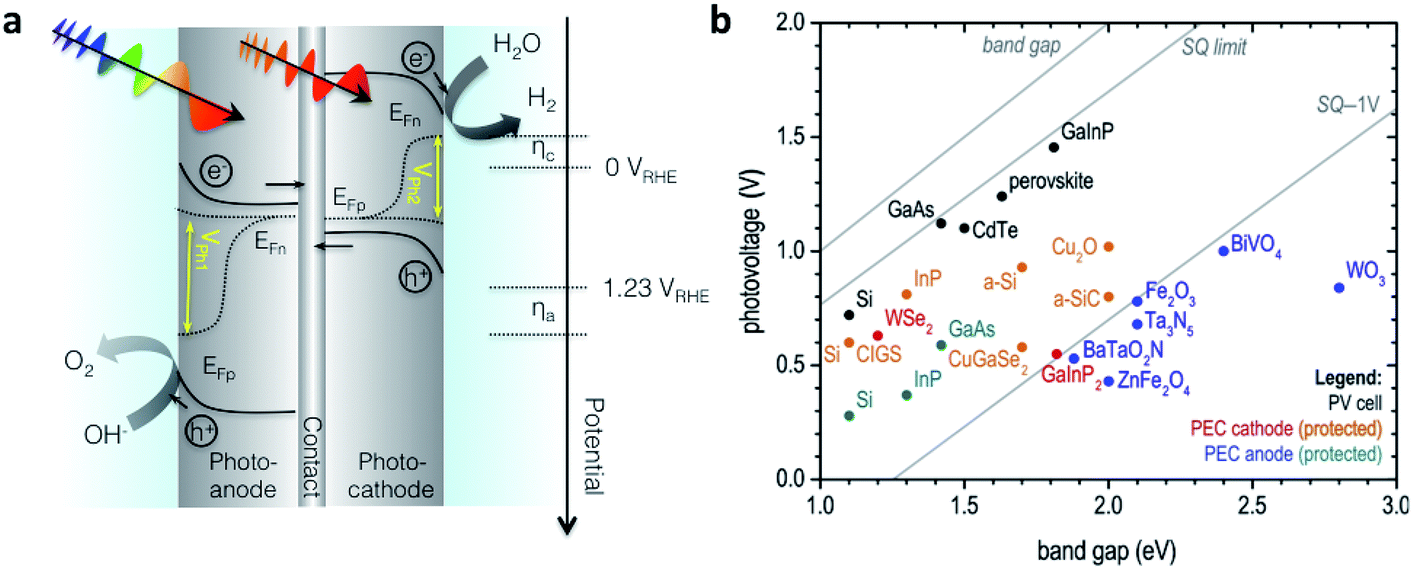 | ||
| Fig. 1 (a) Device architecture of a PEC cell based on an inorganic semiconductor/electrolyte junction. A wide band gap semiconductor photoanode and a lower band gap semiconductor photocathode provide enough photovoltage (VPh1 + VPh2) to drive water splitting. (b) Photovoltage benchmarks for PEC and PV materials as a function of optical band gap. PV devices are plotted by VOC values from devices of top reported efficiency. Vph values for PEC electrodes are taken from published onset potentials, with photocathodes referenced to EHER° (0 VRHE), and photoanodes referenced to EOER° (1.23 VRHE). Diagonal lines represent the band gap, the Shockley–Queisser (S–Q) photovoltage limit, and the S–Q limit minus 1 V. Devices are color-coded to denote those employing buried junctions. Fig. 1b is adapted from ref. 6 with permission from Elsevier. | ||
Although these strategies improve photovoltage generation in these photoelectrodes, their full potential has still not been exploited due to the inherent photovoltage limitation of the semiconductor/electrolyte solution junction. In order to achieve photovoltages closer to the Shockley–Queisser limit, the redox couple has to be carefully chosen with respect to the Fermi level of the semiconductor to maximise the built-in voltage, and the reaction has to be on timescales that are competitive with charge recombination in the material. On the other hand, the implementation of well-designed buried pn junctions into photoelectrode designs significantly improved photovoltage generation and utilisation (Fig. 1b).6 This approach is adapted from photovoltaics where pn junctions are carefully engineered and combined with suitable charge selective contacts to minimise photovoltage losses through recombination. Coupling such a buried pn junction to suitable electrocatalysts lifts the photovoltage limitation in a semiconductor/liquid junction type device and allows for the utilisation of photovoltages close to the Shockley–Queisser limit. Furthermore, through the spatial separation of electrons and holes in such a buried pn junction type photoelectrode charge carrier lifetimes are significantly prolonged to reach timescales required for catalysis. However, using traditional inorganic materials from the photovoltaic field, the design of suitable pn junctions is often assimilated with careful engineering of a defect-free junction using high quality materials (e.g. III–V compound semiconductors) that provide sufficiently long carrier diffusion lengths to reconcile maximum light absorption with efficient charge extraction.1 These materials are significantly limited by cost, however, and there are currently only a few examples of buried junctions using low-cost inorganic semiconductors such as Cu2O as photoabsorbers.15,16
On the other hand, engineering a donor–acceptor junction with organic semiconductors can be achieved at low-cost via established large-area printing techniques for the fabrication of organic photovoltaics (OPVs). In the following we will introduce the concept of a bulk heterojunction (BHJ), before we outline how it entered the field of OPEC water splitting cells.
Bulk heterojunction organic photovoltaics
Organic semiconducting materials are based on molecular frameworks of sp2 hybridised carbon atoms, whereby the extended delocalisation of pi-electron orbitals between neighbouring atoms results in the formation of an effective bandgap Eg between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). In highly conjugated molecular or polymeric systems, this bandgap is small enough such that an electron can be photoexcited from the HOMO to the LUMO upon absorption of incident solar photons, leaving behind a hole to form an electron–hole pair (exciton). Organic semiconductors benefit from excellent light harvesting properties due to their large extinction coefficients that arise from the significant overlap between the ground and excited states of the molecule, as well as their broad and tuneable absorption bands, which can be tailored through chemical design to match the incident solar spectrum. These properties, combined with the low-cost, relative earth abundance and solution processability of organic semiconductors, makes these materials excellent candidates for light harvesting in PEC cells as well as organic photovoltaic applications.Photoelectrodes based on single organic semiconducting materials such as polyaniline or polythiophenes were investigated in the past, but these gave very low photocurrent densities on the order of a few microamperes per square centimetres, which can be partly explained by the large exciton binding energies in organic semiconductors.17,18 Due to the relatively low dielectric constant of organic semiconductors (εr ∼ 2–4), the large Coulomb attraction between charges is poorly screened. This results in high rates of charge recombination following photoexcitation, which reduces the available photocurrent. In order to overcome this problem of charge recombination, the OPV community in the 1980s developed an approach of combining two different organic semiconductors with offset HOMO and LUMO energy levels, such that an electron which is photoexcited in the material with smaller electron affinity (the ‘donor’) is transferred to the second material (the ‘acceptor’), which facilitates charge separation at this interface as demonstrated in Fig. 2a.19 Since the exciton diffusion length in organic semiconductors is only around 10 nm,20 these two materials are typically blended on the nanoscale in order to increase the donor–acceptor interfacial area, creating what is known as a bulk heterojunction (BHJ) OPV device.21,22
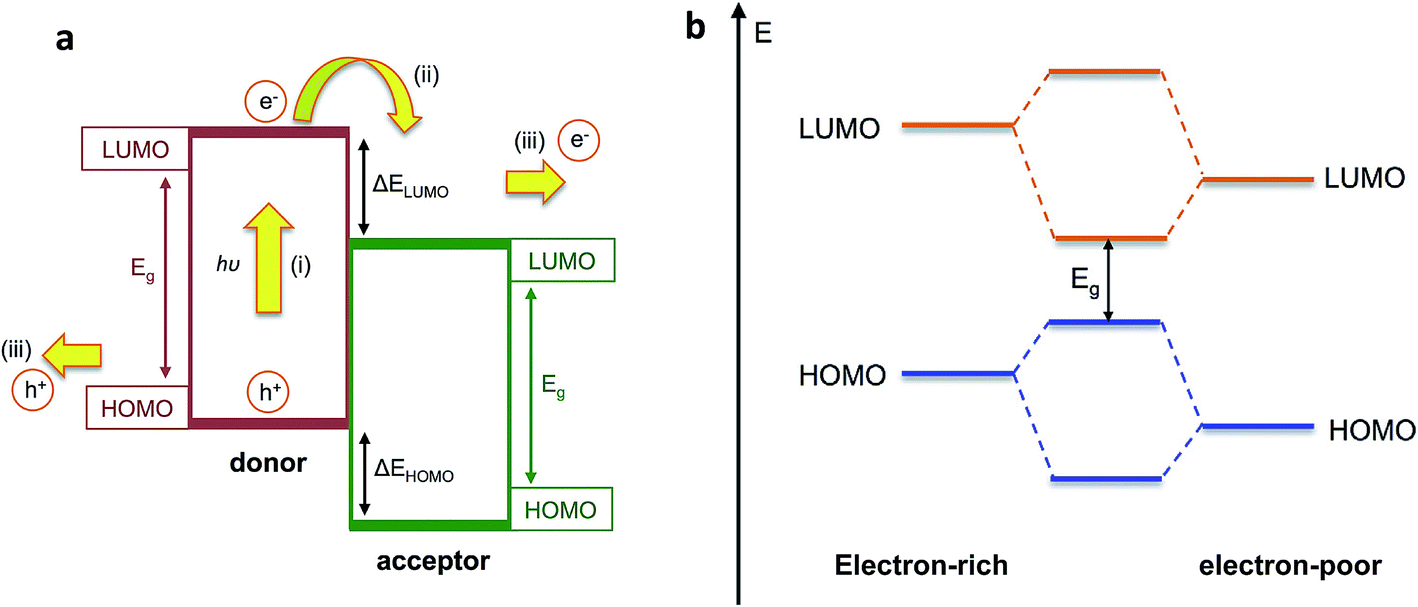 | ||
| Fig. 2 (a) Simplified energy level diagram for photoexcitation and electron transfer in bulk heterojunction blend indicating (i) photoexcitation of an electron into the LUMO of the donor to form an exciton; (ii) electron transfer to the LUMO of the acceptor and (iii) transport of both charges to be collected at the electrodes; (b) reduction in bandgap Egvia molecular orbital hybridisation in push–pull polymers. Adapted from Holliday et al., Prog. Polym. Sci., 70, 2017, 34–51, with permission from Elsevier.23 | ||
The performance of organic semiconductors is determined by many different factors that can be controlled during the design and synthesis of the polymer or small molecule. For example, the planarity of the semiconducting backbone, which controls both the degree of conjugation and crystal packing of the material, can be modified by increasing the number of fused rings or bridging atoms between rings. In addition, the judicious choice of electron-withdrawing (fluorine, carboxylate, imide etc.) or electron-donating (thiophene, alkyloxy etc.) functional groups can tailor the frontier energy levels of the molecule and therefore the optical bandgap, as well as the available voltage of the device. In addition to the optoelectronic properties, device performance is equally dependent on the morphology of the interpenetrating network of donor and acceptor materials within the blend. As such, the density and length of solubilising side-chains on the organic semiconductor can be tuned in order to optimise the phase behaviour and crystal packing of each material, which in turn can affect the charge generation and transport properties of the bulk heterojunction. Finally, the molecular weight properties and purity of the material can be strongly affected by the polymerisation conditions, which in turn can influence the device performance.
One of the earliest and most widely studied donor polymers in the field of OPV is the simple homopolymer poly(3-hexylthiophene) (P3HT) (Fig. 6). Typically it is blended with the fullerene derivative phenyl-C61-butyric acid methyl ester (PCBM) as the acceptor (see Fig. 7), giving a power conversion efficiency of 3–5% in OPV devices.24 The well studied nature of this blend within the OPV community, combined with its reasonably high hole mobility25 and demonstrated aqueous stability,26 made this an obvious choice for early organic photocathode studies. Indeed, the vast majority of organic photoelectrode studies to date have focused on these P3HT:PC61BM blends, as discussed in more detail below.
The rise of organic photoelectrochemical cells
Organic semiconductor/electrolyte junction versus buried junction architectures
In the early stages of research, OPEC water splitting in aqueous solution was demonstrated with polymer photocatalyst/electrolyte solution junctions, yielding photocurrent densities on the order of only a few microamps or less.17,27,28 Striving for higher current output, polymer absorbers were combined with known hydrogen evolution reaction (HER) catalysts – a known strategy from PEC water splitting with inorganic materials. While p/n bilayer junction designs still yielded only a few tens of microamps of photocurrent, even with Pt surface modification,29 the breakthrough leap in photocurrent densities from the microamp to milliamp scale was achieved introducing BHJs into the photoelectrode design and along with HER catalysts and charge selective layers. Bourgeteau et al. first introduced the PEDOT:PSS/P3HT:PCBM/TiO2:MoSx photocathode design (Fig. 3a), inspired by the idea of TiO2 electron extraction layers in the OPV field.30 With the aim to protect the P3HT:PCBM blend from oxygen and water, they developed a thick catalyst layer of MoSx intermixed with TiO2 nanoparticles that could achieve photocurrent densities of 180 μA cm−2 at 0 V vs. RHE and a photocurrent onset potential for hydrogen evolution of ∼0.45 V (Fig. 3b). The photovoltage achieved in the OPEC device was increased by addition of TiOx, approaching the open circuit potential (VOC) of the equivalent OPV device. However, the observed photocurrent densities from hydrogen evolution were more than 50× smaller than the short circuit current obtained in the OPV device.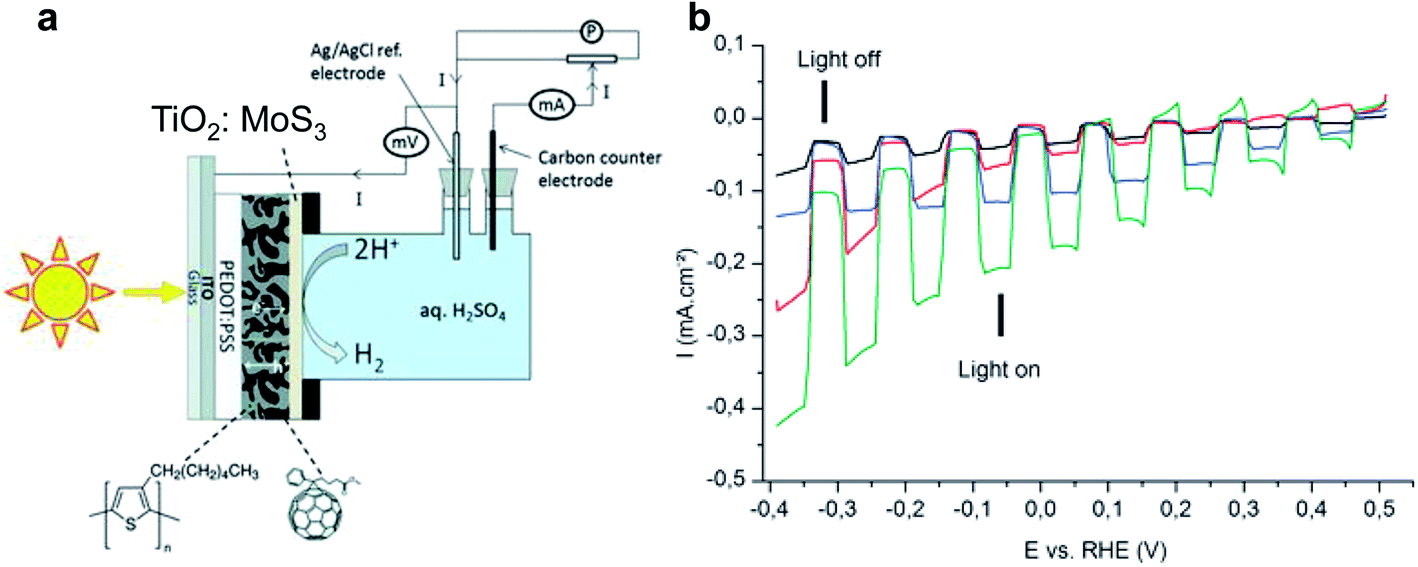 | ||
| Fig. 3 (a) Architecture of the MoS3:TiO2/P3HT:PCBM H2-evolving photocathode inserted in the cell used in this study for photo-electrochemical measurements. (b) Linear sweep voltammograms recorded at 5 mV s−1 in H2SO4 0.5 M with chopped visible light for several photocathodes. Black: P3HT:PCBM (reference), red: MoS3/P3HT:PCBM photocathode, blue: TiO2/P3HT:PCBM photocathode, green: TiO2:MoS3/P3HT:PCBM photocathode. Electrode area: 0.5 cm2. Reproduced from ref. 30 with permission from The Royal Society of Chemistry. | ||
In a subsequent study, Bourgeteau et al. developed an OPEC photocathode that successfully extracted the full photovoltage and photocurrent of an equivalent OPV device by placing a highly conducting LiF (1.2 nm)/Al (100 nm)/Ti (30 nm) interfacial layer between the BHJ and the HER catalyst (Fig. 4a).31 Using this strategy, they could achieve photocurrent densities of −8 mA cm−2 at 0 V vs. RHE with a noble metal-free OPEC photocathode. It is worth noting that these relatively thick metallic interfacial layers not only provide the necessary conductivity and band alignment to efficiently extract photogenerated electrons from the organic blend to the catalyst, but act also as a mirror to reflect otherwise transmitted photons back into the device. Reflective contacts of 100 nm Ag have been shown to enhance the short circuit current by 1/3 in OPV devices.32 Transmission losses should therefore be considered when optimising OPEC photoelectrodes, as they can account for large losses in photocurrent.33 This work also highlights that light harvesting and catalytic components can be optimised separately because the photocurrent onset potential for hydrogen evolution is determined by the sum of the onset potential for dark electrolysis (carried out by the electrocatalyst) and the photovoltage of the junction (Fig. 4b). Using a Pt/C electrocatalyst instead of MoSx, the photocurrent onset could be shifted to ∼200 mV more positive potentials due to a smaller overpotential for hydrogen evolution of the Pt/C electrocatalyst (compare the dark current onset potentials during dark electrolysis for Pt/C and MoSx in Fig. 4b). This demonstrates that such a buried BHJ photoelectrode device can be truly competitive against a PV + electrolyser wired system.
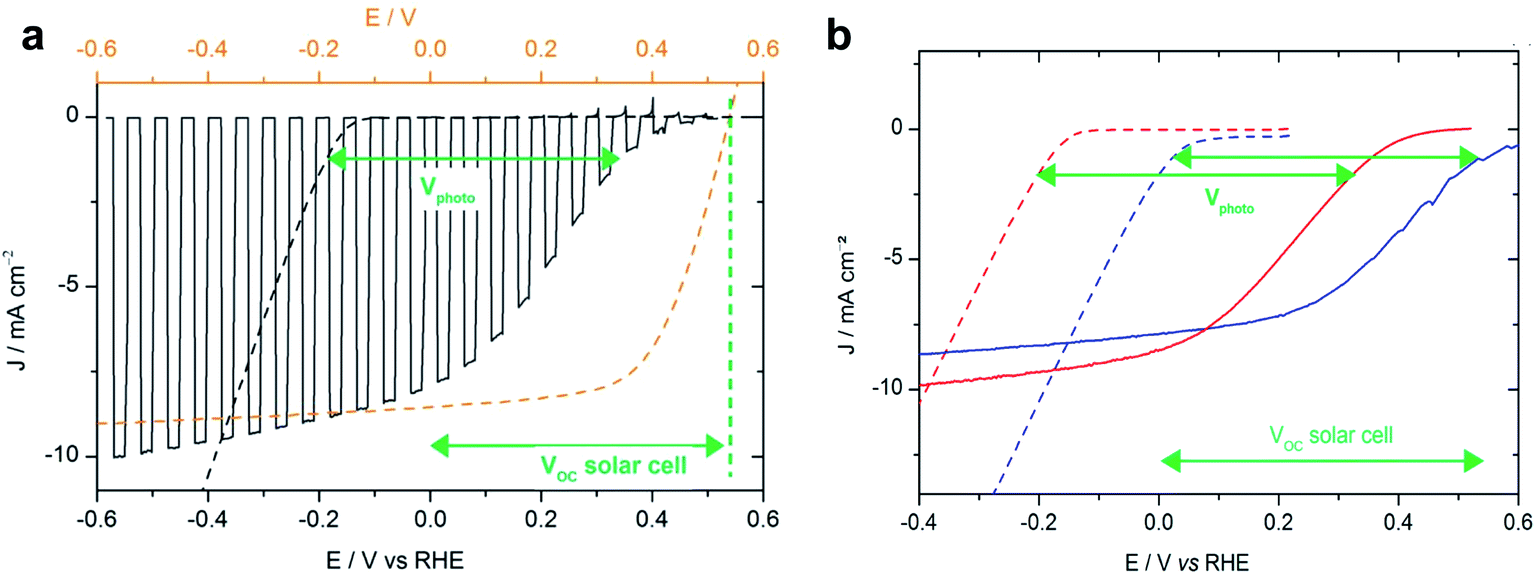 | ||
| Fig. 4 (a) Voltammogram recorded at 50 mV s−1 in 0.5 M H2SO4 with chopped visible light for an ITO/PEDOT:PSS/P3HT:PCBM/LiF/Al/Ti–MoS3 photocathode (black line, electrode area 0.32 cm2) and recorded at 5 mV s−1 in 0.5 M H2SO4 for an ITO–MoS3 cathode (black dashed line, electrode area 0.28 cm2). Potentials are referred to the RHE (bottom axis). The current–voltage curve of an ITO/PEDOT:PSS/P3HT:PCBM/LiF/Al solar cell (orange dashed line, top axis) is shown for comparison. (b) Voltammogram recorded at 5 mV s−1 in 0.5 M H2SO4 for an ITO–MoS3 cathode (red dotted line) and an ITO–Pt/C cathode (blue dotted line) and at 50 mV s−1 with visible-light illumination (100 mW cm−2) for an ITO/PEDOT:PSS/P3HT:PCBM/LiF/Al/Ti–MoS3 photocathode (red line) and an ITO/PEDOT:PSS/P3HT:PCBM/LiF/Al/Ti–Pt/C photocathode (blue line). Reprinted with permission from Bourgeteau, et al., ACS Appl. Mater. Interfaces, 7, 30, 16395–16403. Copyright (2015) American Chemical Society.31 | ||
Meanwhile, Guerrero et al. studied charge extraction from P3HT:PCBM to a redox couple in acetonitrile solution. Using a ferrocene/ferrocenium redox couple, they showed that all charges generated by the P3HT:PCBM blend in an equivalent OPV device could successfully be extracted to the hole scavenger in solution for this particular OPEC device.32 This work might suggest that a buried junction approach using charge selective layers and HER or OER catalysts may not be required for efficient charge extraction in OPEC water splitting cells. However, it is important to note that charge extraction from such an OPEC cell using a direct BHJ-solution interface can only be efficient as long as the electrolyte solution is able to regenerate the oxidised photoabsorber (in this case P3HT) before charge recombination within the BHJ annihilates the oxidised excited state. The language of “regeneration” in this case comes from the picture of a photoelectrochemical dye-sensitised solar cell (DSSC), wherein an electron is injected from the LUMO of the excited dye molecule to the conduction band of TiO2.34 The redox couple in the electrolyte then regenerates the oxidised dye. The open circuit voltage, VOC, will depend solely on the redox potential of the electrolyte and the flat band potential of the TiO2 but such a PEC cell can only function when this redox potential is more negative (higher in energy) than the HOMO of the dye. This picture can be adapted to the PEC cell Guerrero et al. used where P3HT is the analogue to the dye molecule and PCBM/ZnO electron acceptors to the TiO2 (Fig. 5). While such a cell can reach relatively high efficiencies, as has been demonstrated for DSSCs, it should be highlighted that these systems may suffer from known charge recombination losses especially if “regeneration” is not fast enough. The Fc/Fc+ redox reaction is a 1-electron transfer and not proton coupled; hence it is suitable for such a device. However, water splitting is a much more complex multi-electron reaction and is therefore much slower. This challenge of charge extraction via a slow reaction is even more severe in single organic semiconductor photoelectrodes. An example gives the work of Bornoz et al. on a poly[benzimidazobenzophenanthroline] (BBL) photoanode.35 Using sulfite as a sacrificial hole acceptor they could obtain photocurrent densities on the order of 230 μA cm−2 while ten times lower photocurrent densities were extracted from direct water oxidation. Even with a sacrificial agent, the obtained photocurrent densities were rather low which is likely due to limitations in charge separation in the BBL. Though the exciton binding energy in BBL is relatively low (E = 0.29 eV, εr = 8.3),36,37 the generation of free charges is still not barrier-free.
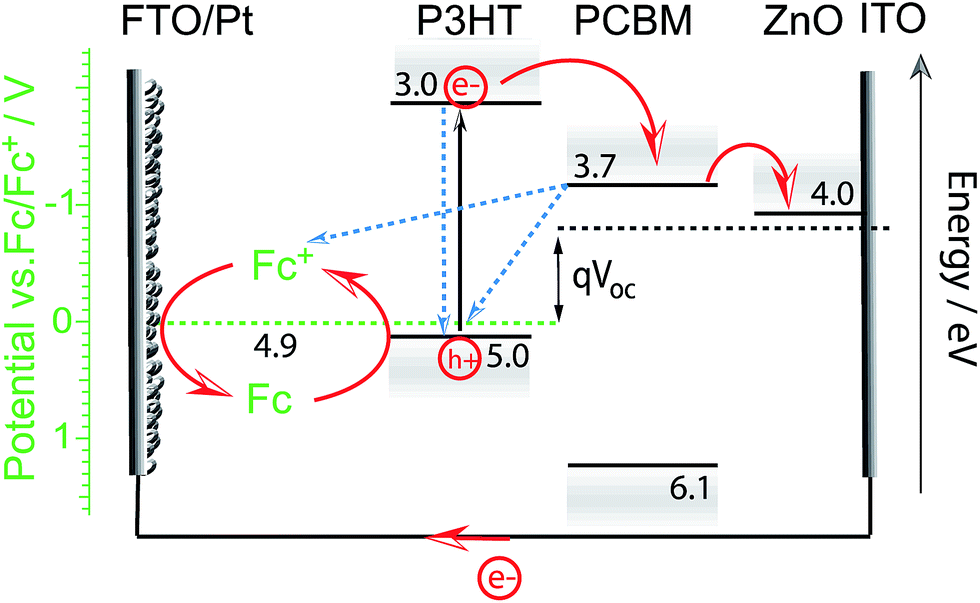 | ||
| Fig. 5 Proposed energy diagram for the photoelectrochemical cell used in the work of Guerrero et al.32 Upon illumination electrons are transferred from P3HT to the acceptor molecule PCBM and through the electron selective contact (here ZnO) to be then extracted to the external circuit and used for the reduction of ferrocenium ions (Fc+) at the FTO/Pt cathode. Electrons from ferrocene (Fc) in the electrolyte regenerate the excited oxidised state of P3HT to close the circuit. The efficiency of the PEC cell will depend on the regeneration efficiency (reaction rate) and recombination rates (indicated by blue dashed arrows). The open circuit potential is determined by the difference in redox potential and Fermi level of the electron selective contact. | ||
Another limitation of a direct semiconductor/liquid interface is that the photovoltage is governed by the redox potential of the electrolyte as shown in Fig. 5. Hence in case of photoanodes for water oxidation, the HOMO level of the donor has to be tailored to the redox potential of the electrolyte, which still remains challenging for materials chemists as we will discuss in the section of low band gap donor polymers.
Additional requirements such as the chemical stability of the organic semiconductor in the electrolyte medium, resistance against the formation of unwanted surface states (e.g. dangling bonds) and stabilised adsorption of both reactants and reaction intermediates at the semiconductor surface must also be considered in this case. In particular, surface adsorption will certainly be an issue that is encountered when using hydrophobic organic semiconductors in aqueous electrolytes. With these limitations in mind, the outlook proposed herein is for the OPEC field to focus on buried BHJ photoelectrode designs that make use of a charge selective layer in-between BHJ and catalyst in order to eliminate the recombination pathway from the blend to solution.
Stability
Since these first demonstrations of efficient proton reduction with organic photocathodes, the OPEC community has focused on the buried junction approach. Interestingly, the reasoning behind the buried BHJ approach was based on introducing an anti-corrosion layer (to protect the blend and PEDOT:PSS) rather than to provide a recombination-blocking layer as we propose is important to minimise charge recombination between the blend and the electrolyte solution.30 Indeed, many of the hole selective layers (HSLs) explored in OPEC cells are corrosion-sensitive (e.g. PEDOT:PSS, polyaniline, MoO3, CuI).26,38–40 For example, MoO3 is commonly used as HSL in OPV devices but encounters problems with intercalation and irreversible reduction into sub-stoichiometric phases of MoO3−xOHx and MoO2, which have unfavourable energy alignment. Hence the strategy of using oxide protective layers was a logical adoption from the inorganic PEC water splitting field, which is essential when using corrosion-sensitive materials such as Cu2O.15 Promising recent examples of oxide protective layers for OPEC devices include the atomic layer deposition (ALD) of amorphous TiO2 onto P3HT:PCBM photocathodes with a CuI HSL, which showed constant hydrogen evolution for more than 3.5 h.41 This was a significant improvement on previous results using amorphous TiO2 deposited by pulsed layer deposition (PLD).40 Thus far, there are only few examples of photoelectrodes showing operating stability on the order of several hours (Table 1). Operational stabilities of up to 3 h for P3HT:PCBM based OPEC devices have been achieved using cross-linked PEDOT:PSS as the hole selective layer and solution-processed TiOx, however the photocurrents measured in this case were extremely low.42 So far, the most promising HSL materials in terms of stability are NiO and WO3, which are able to sustain milliampere range photocurrents over the course of up to 10 h.43,44| HSL | Blend | ESL | Catalyst | Electrolyte | Potential [V vs. RHE] | Stability [hours] | Ref. |
|---|---|---|---|---|---|---|---|
| x-PEDOT (sp) | P3HT:PCBM | TiOx (sp) | Pt (PVD) | 0.1 M Na2SO4 (pH 2) | 0 | 3 | 42 |
| CuI (sp) | P3HT:PCBM | a-TiO2 (ALD) | RuO2 (e-dep) | 0.1 M Na2SO4 (pH 1.36) | 0 | 3.5 | 41 |
| WO3 (PVD) | P3HT:PCBM | a-TiO2 (PLD) | Pt (PVD) | 0.1 M H2SO4 (pH 1.37) | +0.2 | 10 | 43 |
| NiO (sp) | PCDTBT:PCBM | a-TiO2 (ALD) | RuO2 (sp nps) | 0.1 M phosphate buffer (pH 7) | 0 | 5 | 44 |
While the use of protective coatings has proven effective to some extent, it is important to consider that relying on inherently corrosion-sensitive materials in PEC water splitting cells is not favourable for large-scale production, since any imperfections in the protective layer (pinholes, cracks, etc.) would lead to increased production costs due to high failure rates, reducing the economic viability of the entire production process.
In order to achieve standalone efficient water splitting with durable OPEC devices, it is therefore important to explore new materials and architectures that can provide improved performance and stability. Fortunately, there is a vast source of literature on high performance organic semiconductors and devices from the organic photovoltaics (OPV) community that can be adapted to OPEC applications. In the following, we will discuss recent progress in OPV materials, including the emergence of low-bandgap polymers and non-fullerene acceptors for improved light absorption and photovoltage, as well as new tandem device architectures that could be applied to organic PEC devices.
Thus far, one promising recent study by Francàs et al. explored the low bandgap polymer PCDTBT in an organic photocathode blended with PCBM and achieved impressive operational photocathode stabilities of >5 h, as well as exhibiting significantly higher photovoltages compared to P3HT:PCBM blends. This demonstrates the huge potential to combine the progress in materials design and device engineering between the fields of OPV and PEC research, in order to develop high efficiency, stable and low-cost OPEC devices.
Materials advances in organic semiconductors: from OPV to OPEC devices
Low-bandgap donor polymers
Because of the limited visible wavelength absorption of fullerene acceptors, much of the focus of OPV materials development has been to design lower bandgap donor polymers in order to harvest more of the solar spectrum. Initial efforts in this area focused on strategies to planarise the molecular backbone and thereby improve the π-orbital overlap. This included the introduction of bridging atoms such as fluorene and indacenodithiophene, fused ring systems such as benzodithiophene and thienothiophene, and increasing the degree of quinoidal character through replacing phenyl ring systems with thiophene-based systems. In more recent years, the strategy of push–pull hybridisation has been widely adopted, whereby the backbone is composed of alternating electron-rich and electron-poor moieties which results in molecular orbital mixing of these components and a reduced effective bandgap, as demonstrated in Fig. 1b.23,45–47 Using the diverse toolkit of synthetic organic chemistry to tune the energy levels of the molecule, this strategy has proven extremely successful in extending the absorption of donor polymers into the near-IR region of the spectrum, with bandgaps below 1.3 eV having been reported.48Meanwhile, the maximum available open circuit from the OPV cell is determined by the energetic offset between the HOMO of the donor and the LUMO of the acceptor, and can therefore be increased by designing donor polymers with low-lying HOMO energies, some examples of which are shown in Fig. 6. The paradox of trying to maximise light absorption by reducing the bandgap, whilst maximising the voltage by trying to lower the HOMO, has been one of the main dilemmas in materials design for OPV electron donor polymers. In the context of OPEC cells, the large voltage of 1.5 V that is needed to drive unassisted water splitting means that it may be necessary in this case to compromise on the photocurrent in order to maximise photovoltage, meaning that a deep HOMO is more of a priority in these applications. Stabilisation of the HOMO in push–pull polymers can be achieved by various strategies such as incorporating electron-withdrawing substituents along the backbone, as exemplified by the development of the fluorinated PTB7 polymer by Liang et al., which has an ionisation potential of 5.15 eV compared to 5.04 eV for the unfluorinated counterpart.49,50 It should be noted that fluorination is a particularly useful strategy in this regard since it typically results in stabilisation of both the HOMO and LUMO, thereby allowing for a narrow bandgap to be maintained whilst increasing the available voltage.51–53 However, it should also be noted that a sufficiently high-lying LUMO on the donor is required to provide an energetic driving force for charge transfer to the acceptor LUMO. Further stabilisation of the donor HOMO was achieved by replacing the electron-donating alkoxy side chains of PTB7 with alkylthienyl side chains in PTB7-Th, also known as PCE-10, resulting in an increased VOC from 0.76 V to 0.81 V.54 A larger ionisation potential of 5.44 eV was then obtained by incorporating thioalkyl chains on the alkylthienyl groups, resulting in 0.89 V with fullerene-based acceptors.55 Similarly, Li et al. recently published the benzodithiophene based polymer PDTB-EF-T, wherein a deep HOMO of −5.5 eV achieved through the synergistic electron-withdrawing effect of the fluorine groups on the alkylthiophene side-chains and ester groups on the thiophene co-monomers.56 In this example, side-chain engineering was used to optimise the interchain interactions and achieve a favourable morphology, resulting in an outstanding fill factor of 0.76 and power conversion efficiency of 14.2%. The medium bandgap benzodithiophene-co-benzodithiophenedione based push–pull polymer PBDBT developed by the Hou group, (also known as PCE-12) has also demonstrated excellent performance when spectrally matched with low bandgap non-fullerene acceptors (as discussed below). The large ionisation potential (5.33 eV) of this polymer in particular may offer promise for high photovoltage OPEC applications.57 Thirdly, a number of oligothiophene-based push–pull polymers have been developed by the Yan group and others that have strong, temperature-dependent aggregation properties, resulting in the formation of robust, near-ideal morphologies with small but phase-pure polymer domains.58–61 Although this class of polymers present challenges in reproducibility and scale up due to the high-temperature solution processing required, many of these polymers such as PffBT4T-2OD and its longer chain analogue PffBT4T-C9C13 deliver excellent photovoltaic performance of up to 13% PCE, with high open circuit voltages of up to 1.1 V that could prove promising for OPEC applications.62
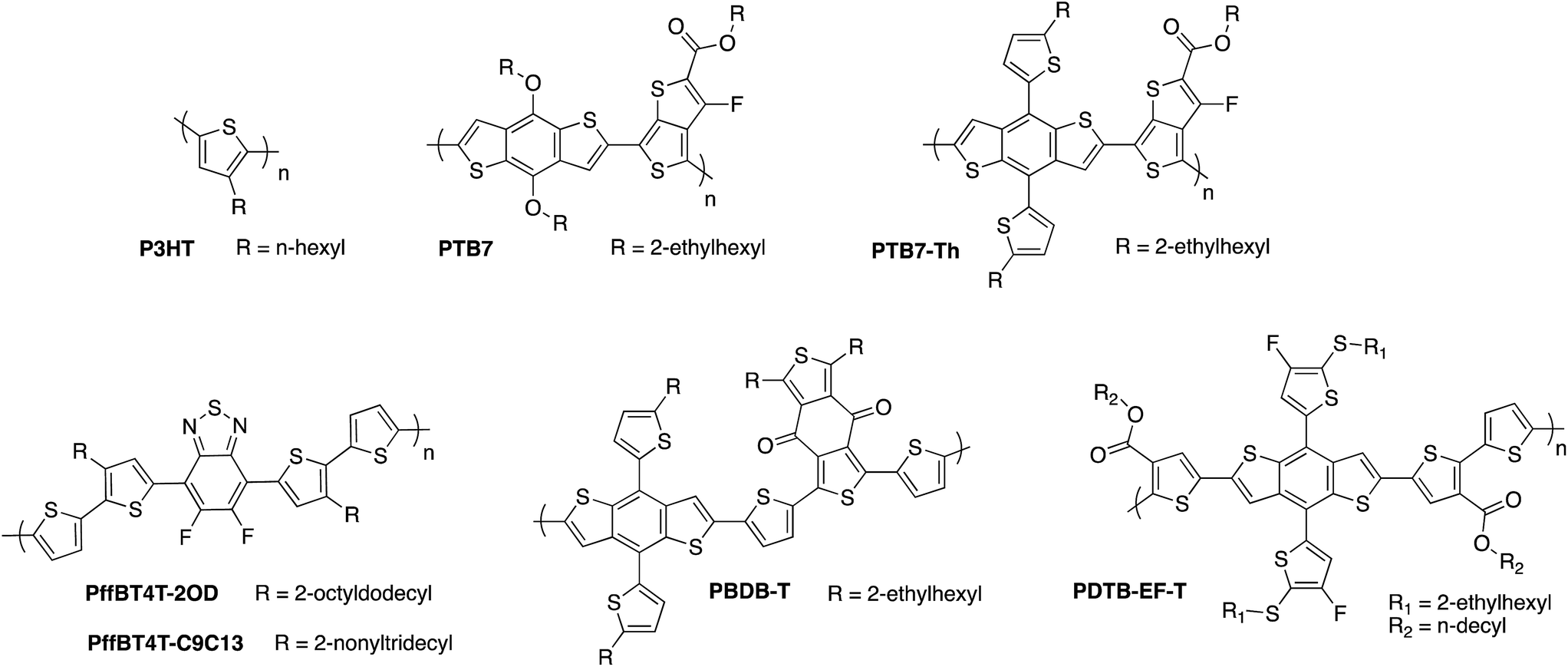 | ||
| Fig. 6 Examples of some of the high performance donor polymers employed in organic photovoltaics research. | ||
As well as selecting materials to maximise photocurrent and photovoltage in photoelectrode applications, it is equally important to consider the stability of these new push–pull polymers in transferring this research to PEC applications. Unfortunately, it has been shown that many of the high-performance donor polymers suffer from significant degradation under illumination in ambient conditions.63–65 Photooxidation and degradation of the polymer backbone is one factor that contributes to this instability, most notably in the case of BDT-TT polymers,66–68 and it has been shown to be accelerated by the use of high-boiling point solvent additives such as 1,8-diiodooctane in deposition of the active layers.69–71 Another source of instability for OPV devices is related to the electrode or interfacial materials, for example in the use of easily oxidised, low-workfunction metal electrodes72 or the acidic, hygroscopic poly(3,4-ethylenedioxythiophene)-polystyrene sulfonate (PEDOT:PSS) hole transport layer, as discussed above.73 These factors will certainly need to be taken into consideration when transferring this research to OPEC applications, as indeed will the stability of these materials under operation in aqueous conditions, which has not yet been studied.
Non-fullerene acceptors
Until very recently, the vast majority of organic solar cell research utilised a fullerene derivative, such as the archetypal phenyl-C61-butyric acid methyl ester (PC61BM) or its C70 analogue PC71BM, as the electron accepting material. Whilst fullerenes have excellent electron transport properties and large electron affinities, making them compatible with many donor polymers, they also have several limitations. Most significantly, C60 fullerenes in particular have very limited absorption in the visible and near-IR region of the solar spectrum due to their high molecular symmetry, which limits the contribution of the acceptor towards light absorption within the active layer. In addition, the very deep LUMO energy of PC61BM and PC71BM significantly limits the available open circuit voltage. Since the optoelectronic properties of fullerenes cannot easily be modified through changes to the chemical structure, it has therefore only been possible until recently to tune the VOC through designing polymers with deeper HOMO energies, which often contradicts the need for a low optical bandgap as mentioned above.Over the past few years, chemists have made considerable breakthroughs in developing alternative, non-fullerene based acceptors (NFAs) with improved visible wavelength absorption and tuneable frontier energy levels, which have resulted in rapid advances in OPV device performance.74–77 The most successful materials in this regard (some example of which are shown in Fig. 7) are based on the same push–pull hybridisation strategy employed in the design of semiconducting polymers, which allows for precise control over the optoelectronic properties, with the phase separation properties optimised through choice of solubilising side-chains and tuning of the backbone planarity. For example, by replacing the fluorene core moiety of FBR for a more extended and electron-rich indacenodithiophene unit in O-IDTBR, the bandgap can be decreased from 2.14 to 1.68 eV.64,78 Further extension of the core in the form of dithieno-indacenodithiophene, as well as modification of the electron withdrawing end groups, gave an even smaller bandgap of 1.59 eV for the small molecule ITIC.79,80 Recent interest in semitransparent OPV devices has inspired chemists to develop increasingly large, fused ring systems with ultra low bandgaps as demonstrated by the 1.25 eV bandgap of 3TT-FIC81 or IEICO-4F (1.24 eV),82 although it should be noted that the added synthetic complexity of such molecules may be less advantageous to commercial scale-up.
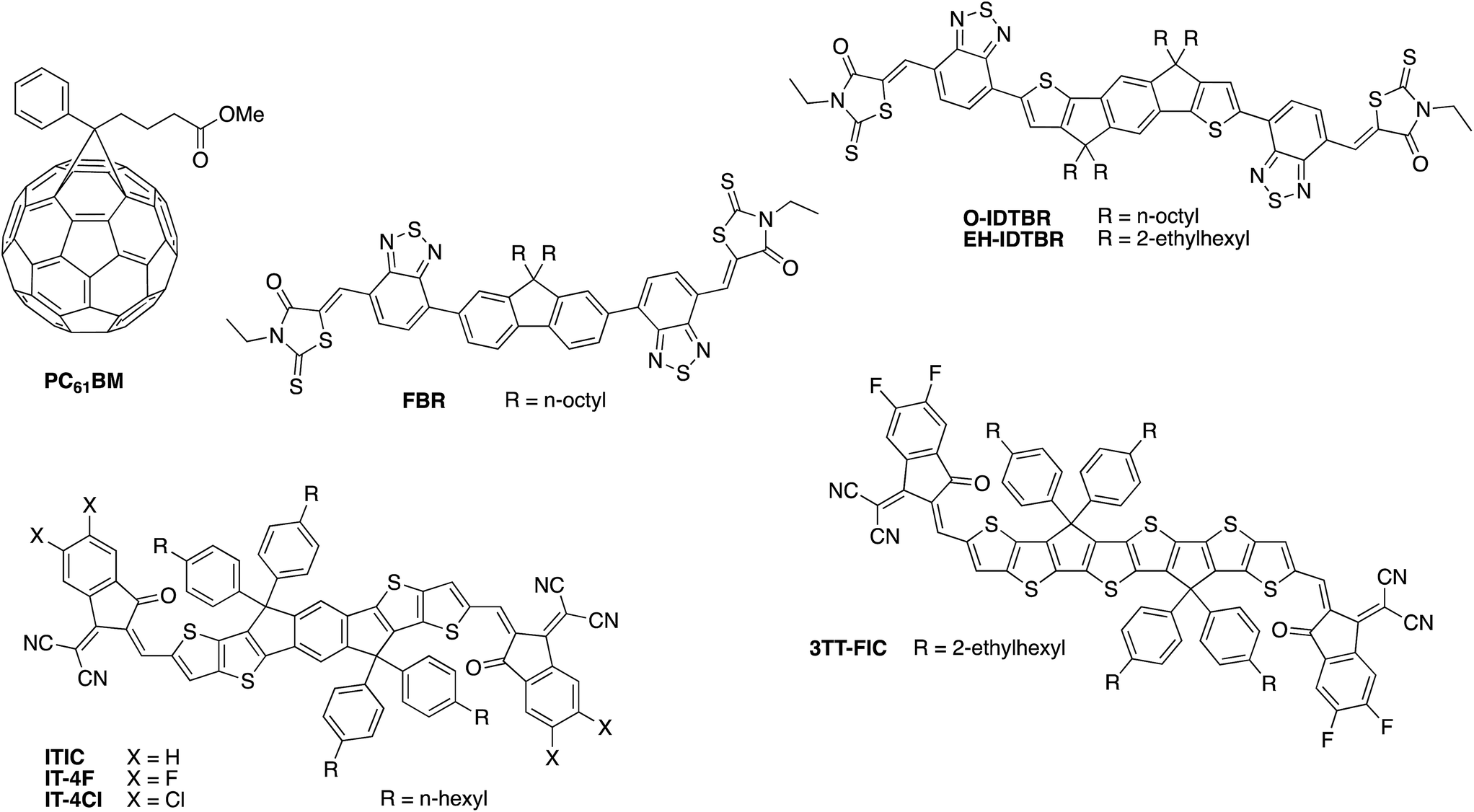 | ||
| Fig. 7 Examples of some of the high performance fullerene and non-fullerene acceptors employed in organic photovoltaics research. | ||
As discussed above, the greater priority in PEC cell applications is to maximise photovoltage in order to have sufficient overpotential to drive unassisted water splitting, in this context, some of the wider bandgap, fluorene-based NFAs may be more promising for OPEC applications, especially when used in conjunction with polymers that have low-lying HOMO energies. For example, the relatively small electron affinity LUMO of FBR (3.57 eV) results in open circuit voltages of up to 1.13 V when combined with the oligothiophene-based polymer PffBT4T-2DT by Baran et al.83 Elsewhere, small molecule NFAs have already delivered many instances of open circuit voltages > 1 V and up to 1.2 V.84 Clearly, the synthetic versatility and degree of optoelectronic control of this new generation of acceptors offers a distinct advantage over their fullerene predecessors. Indeed, the ability to tune both the donor and acceptor simultaneously has successfully liberated the field of OPV from its previous paradigm, whereby the donor polymer was historically responsible for the majority of light absorption as well as maximising the VOC through maintaining a deep HOMO. The opportunity to optimise both materials in the blend has recently led to a greater overall performance in OPV devices, and certainly presents interesting opportunities to explore NFA blends in the context of OPEC devices.
As well as contributing to enhanced photovoltaic performance, non-fullerene acceptors have been shown to offer improved stability in OPV applications.63,64,78,85 This is largely due to the formation of a more stable morphology compared to fullerene-based acceptors, which have a tendency to diffuse through the BHJ blend and aggregate over time.86–88 While many NFA based blends have demonstrated excellent stability in ambient conditions and under illumination, the stability of these blends in aqueous environments has not yet been explored. An important part of the work in transferring this research to the field of PEC cells will therefore require the aqueous stability of these materials to be studied as a first instance, as well as modifying their cohesive and adhesive properties under operation in PEC architectures.
Device architectures and interfacial engineering
Inverted versus conventional architectures
In addition to active layer materials, the performance of an organic photovoltaic device is strongly affected by the choice of device architecture and interfacial layers. In the conventional OPV architecture depicted in Fig. 8, a transparent conducting oxide (typically indium tin oxide) is used as the high work function, transparent anode, with a selective hole transport layer such as the solution processed PEDOT:PSS deposited between the anode and the active layer. A low workfunction metal such as Al is typically used as the cathode in this case, with an additional interlayer of Ca or LiF to facilitate electron injection. This conventional architecture has some disadvantages, in particular with regards to stability due to the reliance on easily oxidised, low workfunction metal electrodes89 and acidic, hygroscopic PEDOT:PSS.90 An alternative, inverted architecture (Fig. 4) was therefore adopted wherein ITO is instead used as the cathode, with an electron transport layer such as ZnO or TiOx solution deposited on top.91,92 MoO3 is often used as the hole selective interlayer in this case and stable metal such as Ag can be used as the top contact, allowing for considerably reduced degradation rates. As well as offering improved stability, the inverted architecture is increasingly used due to the enhanced photovoltaic performance that it offers for many BHJ blends. This may be partly due to the increased light absorption achieved through the use of high reflectance Ag electrodes, but also because of differences in vertical phase separation within the blend. In each configuration, differences in surface energy at the electrode can favour the build-up of a polymer-rich or acceptor-rich layer at the interface. This can significantly affect the charge carrier mobilities and the extent of charge recombination at this interface, which therefore impacts short circuit currents and fill factor, highlighting the importance of tailoring the interfacial properties for enhanced performance.93,94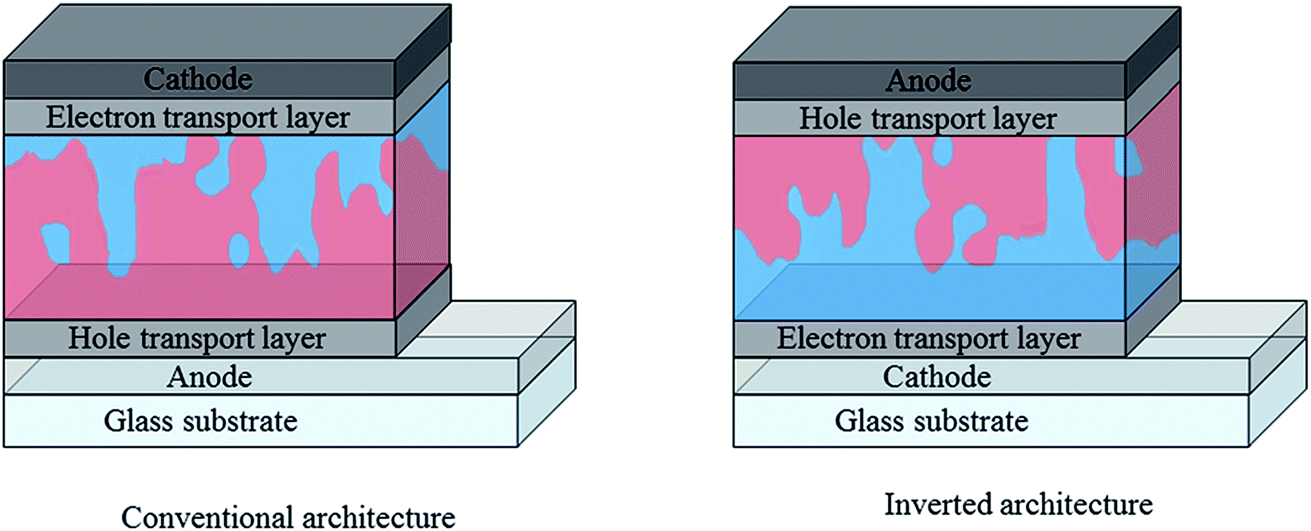 | ||
| Fig. 8 Simplified diagrams depicting conventional and inverted architectures used in single junction organic solar cells; red represents donor-rich regions and blue represents acceptor-rich regions. Reproduced from ref. 76 with permission from the Royal Society of Chemistry. | ||
Since the majority of high performance OPV materials are now optimised for the inverted architecture, one important aspect of developing OPEC devices will be to investigate how the vertical phase separation behaviour and device performance change when using a conventional architecture for photocathodes. At the same time, this will involve investigating alternative charge selective layers that can allow for preferential vertical phase separation within the active layer, whilst improving the interfacial cohesion and stability of the device.
Multijunction device designs
As well as advances in interfacial engineering, the development of high performance OPEC devices can certainly benefit from recent progress in tandem and multijunction architectures as a route to achieving higher photovoltages whilst harvesting more of the solar spectrum.95–97 In single junction OPV devices, the VOC is limited by the bandgap of the donor and acceptor materials, which leads to inherent energy losses due to the thermalisation of high-energy photons (E > Eg). By combining 2 or more sub-cells with complementary bandgaps and connecting them in series, as demonstrated in Fig. 9, the total theoretical VOC becomes the sum of the individual VOCs of the subcells and can often be sufficient to provide the necessary voltage of >1.8 V to drive water splitting. This architecture allows the use of strongly absorbing catalysts and larger electrolyte solution volumes in OPEC devices, since light absorption has only to be optimised within the photocathode. In addition, catalysts with reflective properties could be used to enhance efficiency. It is worth mentioning that although there have been some examples of organic photoanodes,35,98 we think that OPEC cells based on tandem-photocathodes are more promising than a photocathode–photoanode tandem cell. As pointed out earlier, water oxidation is a complex and slow reaction, making efficient hole extraction difficult. In addition, many organic materials are sensitive towards oxygen and suffer from degradation upon exposure. Furthermore, the tandem architectures shown below are good starting points to explore these systems for OPEC water splitting.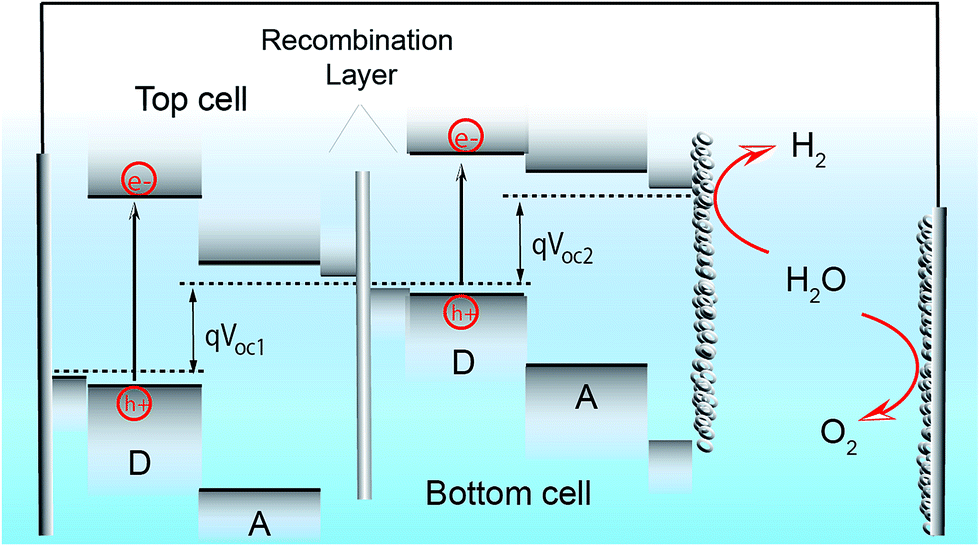 | ||
| Fig. 9 Schematic of a simplified energy diagram of a tandem photocathode comprised of two sub-cells with complementary absorption spectra that are connected in series via a recombination layer and coated with a HER catalyst. A dark electrode is used for water oxidation. This architecture allows the use of strongly absorbing catalysts and larger electrolyte solution volumes between photocathode and anode, since light absorption only needs to be optimised within the photocathode. In addition, catalysts with reflective properties could enhance efficiency. Using a dark anode for oxygen evolution can help to overcome many hurdles in PEC water splitting, such as the susceptibility of many organic semiconductors towards degradation under oxygen exposure. | ||
In addition, by selecting materials with complementary absorption across the spectrum, larger photocurrents can be achieved from multijunction devices due to enhanced spectral coverage (Fig. 10).
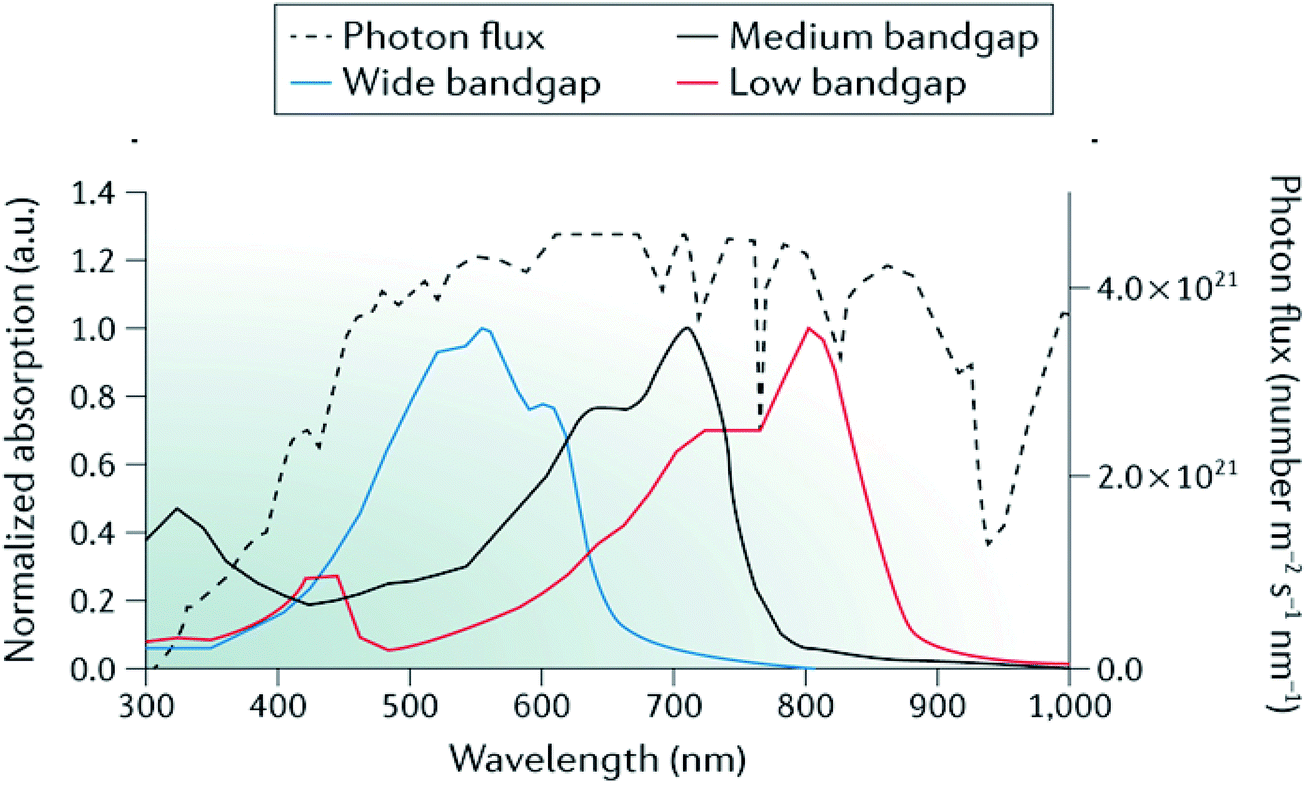 | ||
| Fig. 10 Representative absorption spectra of wide-bandgap, medium-bandgap and low-bandgap donor polymers used in tandem organic solar cells alongside the AM 1.5G solar spectrum (black dotted curve). ICL, interconnecting layer; TCO, transparent conductive oxide. Adapted by permission from Springer Nature: Li et al., Nature Mater. Rev., Copyright 2017.97 | ||
Tandem and triple junction OPV devices have gone through remarkable advances in performance in recent years, accelerated by the emergence of highly tuneable non-fullerene acceptors as well as new medium and wide-bandgap donor polymers, which present new opportunities to tailor the spectral coverage and voltage of the complementary subcells. With regards to OPEC devices, the opportunity to produce much higher photovoltages >1.8 V using multijunction devices is particularly important in terms of meeting the high voltage requirements for water splitting. Since the number of available donor and acceptor materials combinations for organic multijunction devices has multiplied in recent years, the performance of organic multijunction devices is limited more by the availability of charge recombination layers which can deliver sufficiently high photon transmittance and efficient charge recombination, whilst avoiding any damage or degradation of the active layer beneath. The introduction of semitransparent, ultrathin metal layers provided significant advances in this area due to the ability to manipulate the optical field distribution as well as providing balanced current densities between subcells, as demonstrated by the MoO3/Ag/polyelectrolyte layer developed by Zou et al.99 Recently, Che et al. published a tandem OPV device based on a vacuum-deposited small molecule:C70 bottom cell and a solution-processed PTB7-Th:non-fullerene acceptor top cell (Fig. 11) which gave impressive power conversion efficiencies of 15% for relatively large device areas (2 mm2) and open circuit voltages of up to 1.6 V.100 A nearly optically and electrically lossless charge recombination layer was employed, consisting of a layer of Ag nanoparticles adjacent to a vacuum thermal evaporation (VTE)-deposited BPhen:C60 electron-filtering buffer layer, with a hydrophilic capping PEDOT:PSS interlayer deposited on top to protect the BPhen:C60 from damage during solution deposition of the top cell. As well as the exceptionally high PCE, this example offers particular promise for photocathode applications due to its optimal performance in the conventional device architecture, unlike many of the high performance OPV devices in the literature.
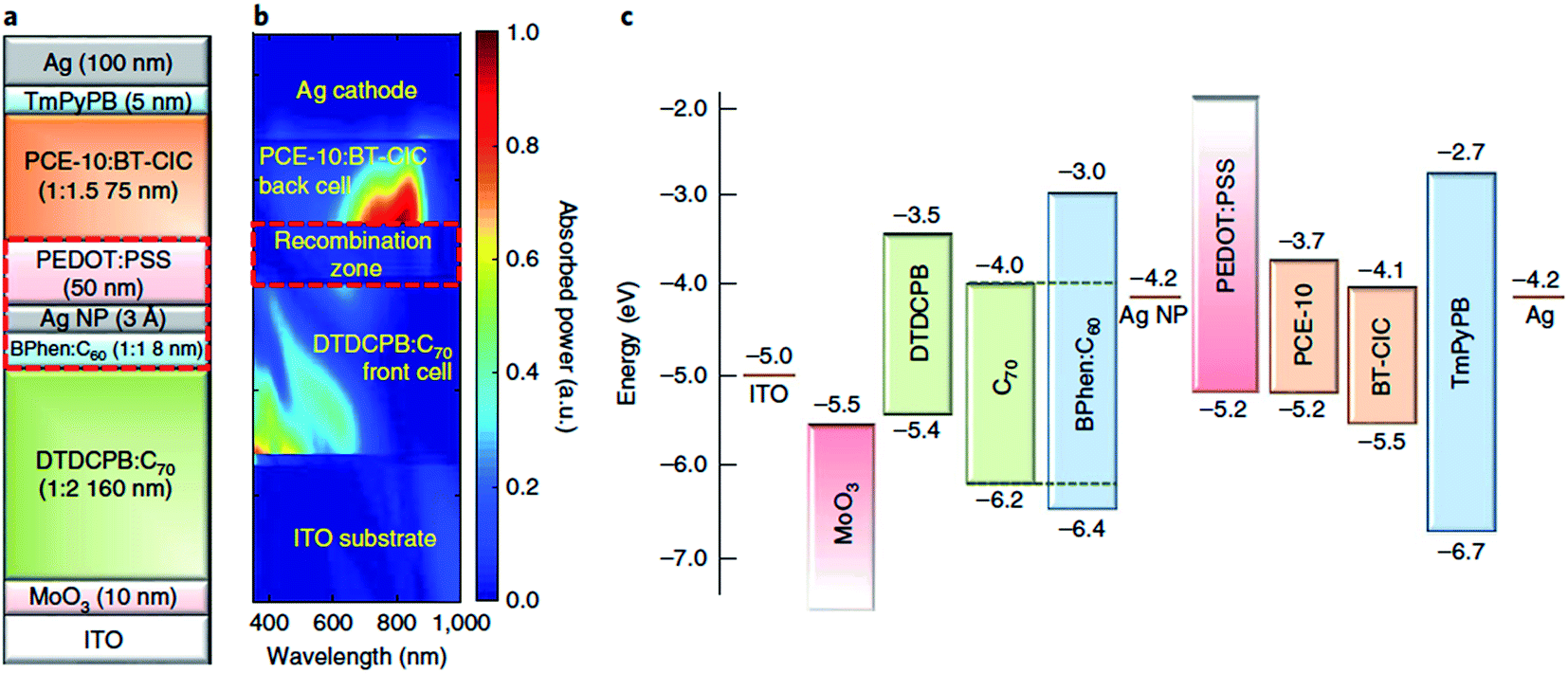 | ||
| Fig. 11 Tandem device architecture published by Che et al. showing (a) schematic of double junction device; (b) simulated absorbed optical power distribution (relative); (c) energy level diagram. Reprinted by permission from Springer Nature: Che et al., Nat. Energy, Copyright 2018.100 | ||
Homo-tandem devices, wherein 2 or more subcells of the same active layer materials are stacked in series or parallel, have also attracted recent interest as a means to enhance the light trapping and photovoltage, as well as offering simplified processing due to the use of only one active layer blend.101,102 Esiner et al. fabricated triple junction OPV devices with 2 identical photoactive layers based on the wide bandgap polymer PTPTIBDT-OD that could produce open circuit voltages up to 1.74 V, which they demonstrated to be large enough to split water in an electrochemical device.103–105 In another example by Chen et al., a double junction homo-tandem device was fabricated with the oligothiophene polymer P3TEA and a perylene diimide based non-fullerene acceptor SF-PDI2, giving an exceptionally high VOC of 2.1 V and 10.8% PCE.106 The high photovoltage of this OPV device was also used to split water in an electrolytic cell, demonstrating the potential to implement homo-tandem devices in photocathodes if the materials are shown to be stable and the design can be adapted to a conventional architecture, buried junction OPEC configuration.
Benchmarking OPEC performance
Finally, with all these new research opportunities it is important to emphasise that progress in the field of OPEC water splitting should be documented in a consistent way. Below we will highlight a few important points that should be considered to make published results more comparable with each other.(1) In order to assess operational stability and faradaic efficiency, OPEC cells should be operated under stated illumination (typically AM 1.5G simulated 1 sun intensity) for several hours whilst simultaneously recording gas production by gas chromatography or Clark electrodes. It is also advisable to carry out these measurements under a nitrogen atmosphere whilst stirring the electrolyte to avoid the build-up of pH gradients and bubble adhesion to the surface of the photoelectrode. The potential applied during the chronoamperometric measurement should be chosen with respect to the reaction measured. For proton reduction, values of 0 V vs. RHE are commonly used, however more positive values can be selected if the photovoltage of the photocathode is sufficiently high. Ideally, these measurements should be performed at the operating potential of an ideal 2-electrode tandem device. For a photoanode-photocathode tandem this potential would be equivalent to the intersection point, Eop, of their Jphoto–V curves as shown in Fig. 12a. In a single-electrode tandem device this would be the intersection point between the Jphoto–V curve of the photoelectrode and the electrolysis curve of the dark electrode.
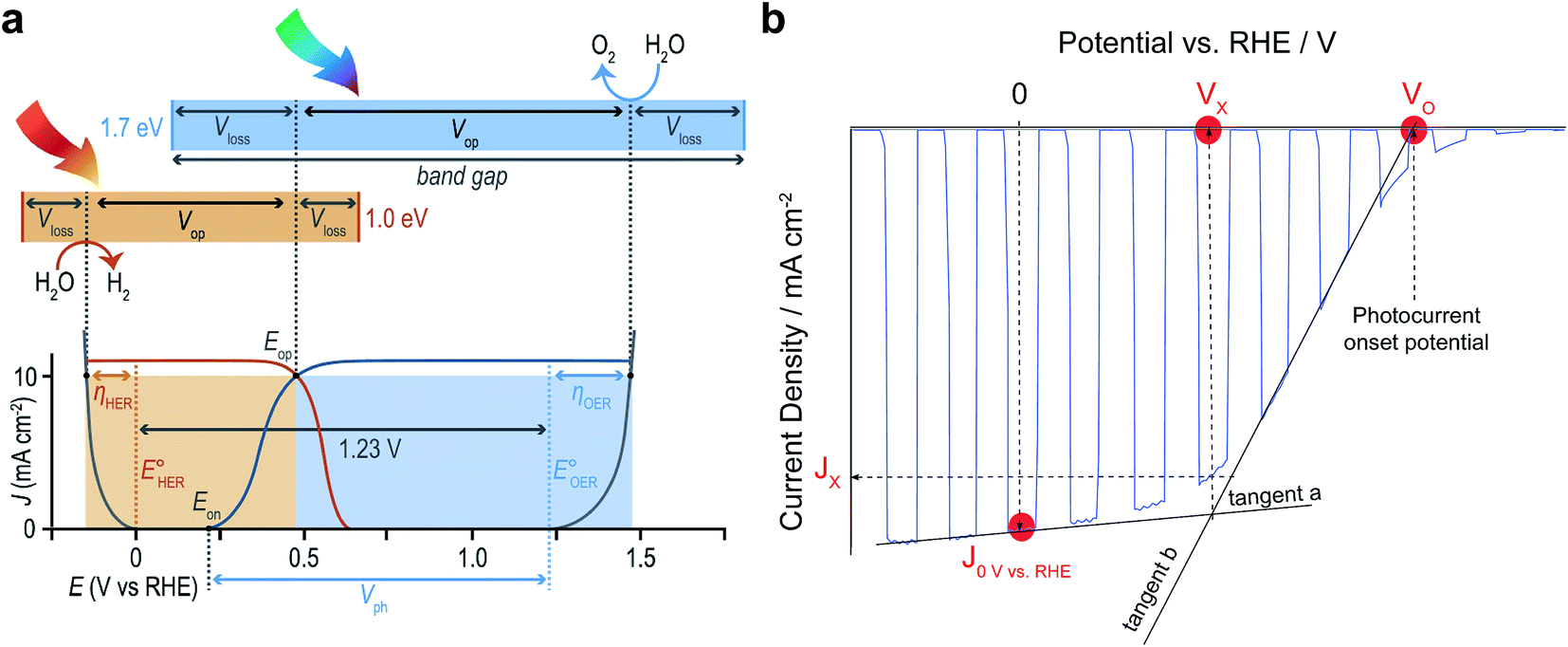 | ||
| Fig. 12 (a) Schematic relating band gaps and photovoltages for an idealised two-absorber tandem photoelectrolysis device in which the anode is the top absorber. Free energy losses in each absorber (Vloss) and catalytic overpotentials for each half-reaction (η) are fundamental loss pathways. Current density–potential curves for electrocatalysts (grey) and an idealised PEC photoanode (blue) and photocathode (orange) are shown with their relative power contributions (shaded areas) in an ideal tandem operating at Eop being the intersection of the two J–V curves. Reprinted from ref. 6 with permission from Elsevier. (b) Useful benchmark parameters in a current density–potential (J–V) curve of typical OPEC cells showing the current density at 0 V vs. RHE, the photocurrent Jx at the cross-over potential Vx and the photocurrent onset potential VO. For simplicity, a photocathode J–V curve was chosen. | ||
(2) When stating photocurrent densities, a comparison at 0 V vs. RHE for photocathodes and 1.23 V for photoanodes has been helpful to compare literature results thus far. It is noted that all J–V curves should be referenced to the RHE scale, taking into account the varying electrolyte solutions and their pH used. Apart from comparing photocurrent densities at these potentials, it is useful to state the photocurrent at the operating potential of an ideal 2-electrode tandem cell as mentioned above. This can be achieved by determining the potential Vx at the intersection of the tangents (a) and (b) as shown in Fig. 12b.
(3) Similarly, the onset potential, VO, for HER or OER can be determined from the intersection of tangent (b) with the potential axis (at zero current). This is a more consistent way of referencing the photocurrent onset potential than determining it by an arbitrary value of 0.1 mA cm−2 as has been proposed earlier.31 Indeed, depending on the catalyst used, some catalysts transform into pre-catalytic species by changing oxidation states. This is usually seen as a redox peak in the linear sweep and might misguide the interpretation of the onset potential. If J–V curves show 2 slopes and it is not clear how to determine the reaction onset potential, it is advisable to measure gas evolution at potentials between these two slopes to confirm faradaic efficiency.
(4) Although the use of noble-metal catalysts is incompatible with a low-cost OPEC cell design, research on novel OPEC BHJs should not be hampered by this criterion. As discussed above, a buried BHJ design offers the advantage to disconnect light harvesting and catalysis components, which means that each component can be optimised separately. However, it is also important to consider the need for mild deposition procedures for earth-abundant catalysts with low overpotentials for HER and OER.
Following these benchmarking guidelines will benefit the OPEC community and help the field to advance more rapidly by unifying the structure and research language within the literature.
Table 2 summarises selected benchmarking performances of OPEC photocathodes. The work of Bourgeteau et al. has achieved the best photocurrent at 0 V vs. RHE (−7.90 mA cm−2). In the work of Rojas et al. the highest photocurrent (−4.84 mA cm−2), at the most positive potential (0.281 V) has been demonstrated. The best photocurrent onset potentials (0.58 V and 0.55 V) have been achieved with PEDOT:PSS/PCDTBT:PCBM/TiO2/RuO2 and CuI/P3HT:PCBM/TiO2/Pt:PEI photocathodes respectively. Though these represent record values, they are not stabilised performances. The ALD-TiO2 protected CuI/P3HT:PCBM/TiO2/RuO2 photocathode shows the best stabilised OPEC performance. Some of the recent examples show rather resistive J–V curves and hence, the cross-over potential, Vx, and photocurrent, Jx, cannot be determined.
| Cathode | V 0 [V vs. RHE] | J 0 VRHE [mA cm−2] | V x [V vs. RHE] | J x [mA cm−2] | Ref. |
|---|---|---|---|---|---|
| a Values could not be determined. | |||||
| PEDOT:PSS/P3HT:PCBM/LiF/Al/Ti/MoSx | 0.39 | −7.90 | 0.053 | −7.42 | 31 |
| CuI/P3HT:PCBM/TiO2/Pt:PEI | 0.55 | −6.78 | 0.281 | −4.84 | 40 |
| CuI/P3HT:PCBM/TiO2/ RuO2 (stabilised) | 0.48 | −3.61 | 0.091 | −3.13 | 41 |
| (100 nm) WO3/P3HT:PCBM/TiO2/Pt (stabilised) | 0.41 | −2.48 | —a | —a | 43 |
| PEDOT:PSS/PCDTBT:PCBM/TiO2/RuO2 | 0.58 | −2.85 | —a | —a | 44 |
| NiO/PCDTBT:PCBM/TiO2/RuO2 (stabilised) | 0.41 | −1.45 | —a | —a | 44 |
Conclusions
In summary, although a direct semiconductor/electrolyte solution junction would be highly desirable for a simple and low-cost PEC design, decades of research on PEC water splitting with inorganic materials has revealed some serious shortcomings of such systems. In particular, these devices suffer from significant photovoltage losses due to water splitting being too slow to compete with charge recombination in the absence of a strong electric field. Buried junction PEC devices have been proven to achieve generally higher photovoltages but are often prohibitively expensive when fabricated with traditional inorganic materials. OPEC water splitting devices, meanwhile, have great potential to realise a cost-effective buried junction type photoelectrode, standing on the shoulders of decade-long research in both the OPV and PEC fields.The rapid advances in materials design within the field of OPV has provided a vast number of high performance donor polymers and non-fullerene acceptors with carefully tailored chemical structures that can now deliver both high photocurrents and open circuit voltages > 1 V. Opportunities to adopt these new materials to OPEC photoelectrodes means that significantly more efficient OPEC devices are now in reach. In addition, the use of improved double or triple BHJ tandem photocathode architectures and interlayer materials could be especially promising for high-efficiency OPEC devices providing photovoltages of >1.8 V necessary for the water splitting reaction.
Finally, with all these new research opportunities it will be important to establish a systematic structure to compare OPEC water splitting cells amongst each other and with their inorganic counterparts, and to easily identify benchmark systems. Towards this goal, we suggest here some benchmarking guidelines and emphasise the need to implement operational stability measurements over several hours into the protocol.
With more research on buried junction OPEC devices implementing some of the most promising blend materials used so far in the OPV field we expect to see rapid advances in OPEC benchmark performances. Simultaneously, the field should move towards reporting stabilised performances. Existing organic blend materials should be screened in order to find candidates for improved stability in aqueous environments under applied potential and light bias. Furthermore, materials design should include synthesis strategies to improve material performance and stability and be tailored to OPEC water splitting needs. Identifying stable and high-performance materials for OPEC photocathodes will pave the way for stable tandem architectures that could demonstrate stable and efficient voltage-unassisted water splitting with low-cost devices.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
L. S. would like to thank the European Research Council (H2020-MSCA-IF-2016 Grant 749231) for funding. S. H. acknowledges funding from the EPSRC (EP/M005143/1 Project 51323). Both authors would like to thank J. R. Durrant and I. McCulloch for proof-reading the manuscript.References
- J. L. Young, M. A. Steiner, H. Döscher, R. M. France, J. A. Turner and T. G. Deutsch, Direct Solar-to-Hydrogen Conversion Via Inverted Metamorphic Multi-Junction Semiconductor Architectures, Nat. Energy, 2017, 2, 17028 CrossRef CAS.
- K. Sivula and R. van de Krol, Semiconducting Materials for Photoelectrochemical Energy Conversion, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- M. S. Prévot and K. Sivula, Photoelectrochemical Tandem Cells for Solar Water Splitting, J. Phys. Chem. C, 2013, 117, 17879–17893 CrossRef.
- F. F. Le, E. Pastor, S. D. Tilley, C. A. Mesa, S. R. Pendlebury, M. Graetzel and J. R. Durrant, Rate Law Analysis of Water Oxidation on a Hematite Surface, J. Am. Chem. Soc., 2015, 137, 6629–6637 CrossRef PubMed.
- F. Le Formal, S. R. Pendlebury, M. Cornuz, S. D. Tilley, M. Gratzel and J. R. Durrant, Back Electron-Hole Recombination in Hematite Photoanodes for Water Splitting, J. Am. Chem. Soc., 2014, 136, 2564–2574 CrossRef CAS PubMed.
- M. T. Mayer, Photovoltage at Semiconductor–Electrolyte Junctions, Curr. Opin. Electrochem., 2017, 2, 104–110 CrossRef CAS.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Light-Induced Water Splitting with Hematite: Improved Nanostructure and Iridium Oxide Catalysis, Angew. Chem., Int. Ed., 2010, 49, 6405–6408 CrossRef CAS PubMed.
- T. W. Kim and K.-S. Choi, Nanoporous BiVO4 Photoanodes with Dual-Layer Oxygen Evolution Catalysts for Solar Water Splitting, Science, 2014, 343, 990 CrossRef CAS PubMed.
- T. Hisatomi, F. Le Formal, M. Cornuz, J. Brillet, N. Tetreault, K. Sivula and M. Gratzel, Cathodic Shift in Onset Potential of Solar Oxygen Evolution on Hematite by 13-Group Oxide Overlayers, Energy Environ. Sci., 2011, 4, 2512–2515 RSC.
- F. Le Formal, N. Tetreault, M. Cornuz, T. Moehl, M. Gratzel and K. Sivula, Passivating Surface States on Water Splitting Hematite Photoanodes with Alumina Overlayers, Chem. Sci., 2011, 2, 737–743 RSC.
- L. Steier, I. Herraiz-Cardona, S. Gimenez, F. Fabregat-Santiago, J. Bisquert, S. D. Tilley and M. Gratzel, Understanding the Role of Underlayers and Overlayers in Thin Film Hematite Photoanodes, Adv. Funct. Mater., 2014, 24, 7681–7688 CrossRef CAS.
- A. Kafizas, X. Xing, S. Selim, C. A. Mesa, Y. Ma, C. Burgess, M. A. McLachlan and J. R. Durrant, Ultra-Thin Al2O3 Coatings on BiVO4 Photoanodes: Impact on Performance and Charge Carrier Dynamics, Catal. Today, 2017 DOI:10.1016/j.cattod.2017.11.
- M. Barroso, A. J. Cowan, S. R. Pendlebury, M. Grätzel, D. R. Klug and J. R. Durrant, The Role of Cobalt Phosphate in Enhancing the Photocatalytic Activity of A-Fe2O3 Toward Water Oxidation, J. Am. Chem. Soc., 2011, 133, 14868–14871 CrossRef CAS PubMed.
- Y. Ma, A. Kafizas, R. Pendlebury Stephanie, F. Le Formal and R. Durrant James, Photoinduced Absorption Spectroscopy of Copi on BiVO4: The Function of Copi During Water Oxidation, Adv. Funct. Mater., 2016, 26, 4951–4960 CrossRef CAS.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Highly Active Oxide Photocathode for Photoelectrochemical Water Reduction, Nat. Mater., 2011, 10, 456 CrossRef CAS PubMed.
- L. Pan, J. H. Kim, M. T. Mayer, M.-K. Son, A. Ummadisingu, J. S. Lee, A. Hagfeldt, J. Luo and M. Grätzel, Boosting the Performance of Cu2O Photocathodes for Unassisted Solar Water Splitting Devices, Nature Catalysis, 2018, 1, 412–420 CrossRef.
- G. Suppes, E. Ballard and S. Holdcroft, Aqueous Photocathode Activity of Regioregular Poly(3-Hexylthiophene), Polym. Chem., 2013, 4, 5345–5350 RSC.
- D. Hursan, A. Kormanyos, K. Rajeshwar and C. Janaky, Polyaniline Films Photoelectrochemically Reduce CO2 to Alcohols, Chem. Commun., 2016, 52, 8858–8861 RSC.
- C. W. Tang, Two-Layer Organic Photovoltaic Cell, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- B. Siegmund, T. Sajjad Muhammad, J. Widmer, D. Ray, C. Koerner, M. Riede, K. Leo, D. W. Samuel Ifor and K. Vandewal, Exciton Diffusion Length and Charge Extraction Yield in Organic Bilayer Solar Cells, Adv. Mater., 2017, 29, 1604424 CrossRef PubMed.
- M. Hiramoto, H. Fujiwara and M. Yokoyama, Three-Layered Organic Solar Cell with a Photoactive Interlayer of Codeposited Pigments, Appl. Phys. Lett., 1991, 58, 1062–1064 CrossRef CAS.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Semiconducting Polymers (as Donors) and Buckminsterfullerene (as Acceptor): Photoinduced Electron Transfer and Heterojunction Devices, Synth. Met., 1993, 59, 333–352 CrossRef CAS.
- S. Holliday, Y. Li and C. K. Luscombe, Recent Advances in High Performance Donor–Acceptor Polymers for Organic Photovoltaics, Prog. Polym. Sci., 2017, 70, 34–51 CrossRef CAS.
- T. Dang Minh, L. Hirsch and G. Wantz, P3HT:Pcbm, Best Seller in Polymer Photovoltaic Research, Adv. Mater., 2011, 23, 3597–3602 CrossRef.
- S. Holliday, J. E. Donaghey and I. McCulloch, Advances in Charge Carrier Mobilities of Semiconducting Polymers Used in Organic Transistors, Chem. Mater., 2014, 26, 647–663 CrossRef CAS.
- F. Fumagalli, S. Bellani, M. Schreier, S. Leonardi, H. C. Rojas, A. Ghadirzadeh, G. Tullii, A. Savoini, G. Marra and L. Meda, et al., Hybrid Organic–Inorganic H-2-Evolving Photocathodes: Understanding the Route Towards High Performance Organic Photoelectrochemical Water Splitting, J. Mater. Chem. A, 2016, 4, 2178–2187 RSC.
- S. F. Yang, L. Z. Fan and S. H. Yang, Significantly Enhanced Photocurrent Efficiency of a Poly(3-Hexylthiophene) Photoelectrochemical Device by Doping with the Endohedral Metallofullerene Dy@C-82, Chem. Phys. Lett., 2004, 388, 253–258 CrossRef CAS.
- E. Lanzarini, M. R. Antognazza, M. Biso, A. Ansaldo, L. Laudato, P. Bruno, P. Metrangolo, G. Resnati, D. Ricci and G. Lanzani, Polymer-Based Photocatalytic Hydrogen Generation, J. Phys. Chem. C, 2012, 116, 10944–10949 CrossRef CAS.
- T. Abe, S. Tobinai, N. Taira, J. Chiba, T. Itoh and K. Nagai, Molecular Hydrogen Evolution by Organic P/N Bilayer Film of Phthalocyanine/Fullerene in the Entire Visible-Light Energy Region, J. Phys. Chem. C, 2011, 115, 7701–7705 CrossRef CAS.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, C. Laberty-Robert, S. Campidelli, R. de Bettignies, V. Artero, S. Palacin and B. Jousselme, A H-2-Evolving Photocathode Based on Direct Sensitization of MoS3 with an Organic Photovoltaic Cell, Energy Environ. Sci., 2013, 6, 2706–2713 RSC.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, R. Cornut, V. Artero and B. Jousselme, Enhancing the Performances of P3HT:PCBM-MoS3-Based H-2-Evolving Photocathodes with Interfacial Layers, ACS Appl. Mater. Interfaces, 2015, 7, 16395–16403 CrossRef CAS PubMed.
- A. Guerrero, M. Haro, S. Bellani, M. R. Antognazza, L. Meda, S. Gimenez and J. Bisquert, Organic Photoelectrochemical Cells with Quantitative Photocarrier Conversion, Energy Environ. Sci., 2014, 7, 3666–3673 RSC.
- P. Cendula, L. Steier, P. A. Losio, M. Gratzel and J. O. Schumacher, Analysis of Optical Losses in a Photoelectrochemical Cell: A Tool for Precise Absorptance Estimation, Adv. Funct. Mater., 2018, 28 CrossRef CAS.
- M. Gratzel, Photoelectrochemical Cells, Nature, 2001, 414, 338–344 CrossRef CAS PubMed.
- P. Bornoz, M. S. Prévot, X. Yu, N. Guijarro and K. Sivula, Direct Light-Driven Water Oxidation by a Ladder-Type Conjugated Polymer Photoanode, J. Am. Chem. Soc., 2015, 137, 15338–15341 CrossRef CAS PubMed.
- S. Kraner, C. Koerner, K. Leo, E. Bittrich, K. J. Eichhorn, Y. Karpov, A. Kiriy, M. Stamm, K. Hinrichs and M. Al-Hussein, Dielectric Function of a Poly(Benzimidazobenzophenanthroline) Ladder Polymer, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 195202 CrossRef.
- S. Kraner, R. Scholz, F. Plasser, C. Koerner and K. Leo, Exciton Size and Binding Energy Limitations in One-Dimensional Organic Materials, J. Chem. Phys., 2015, 143, 244905 CrossRef CAS PubMed.
- E. Belarb, V. M. Blas-Ferrando, M. Haro, H. Maghraoui-Meherzi and S. Gimenez, Electropolymerized Polyaniline: A Promising Hole Selective Contact in Organic Photoelectrochemical Cells, Chem. Eng. Sci., 2016, 154, 143–149 CrossRef.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, S. Campidelli, R. Cornut and B. Jousselme, All Solution-Processed Organic Photocathodes with Increased Efficiency and Stability Via the Tuning of the Hole-Extracting Layer, J. Mater. Chem. A, 2016, 4, 4831–4839 RSC.
- H. C. Rojas, S. Bellani, F. Fumagalli, G. Tullii, S. Leonardi, M. T. Mayer, M. Schreier, M. Gratzel, G. Lanzani and F. Di Fonzo, et al., Polymer-Based Photocathodes with a Solution-Processable Cuprous Iodide Anode Layer and a Polyethyleneimine Protective Coating, Energy Environ. Sci., 2016, 9, 3710–3723 RSC.
- L. Steier, S. Bellani, H. C. Rojas, L. F. Pan, M. Laitinen, T. Sajavaara, F. Di Fonzo, M. Gratzel, M. R. Antognazza and M. T. Mayer, Stabilizing Organic Photocathodes by Low-Temperature Atomic Layer Deposition of TiO2, Sustainable Energy Fuels, 2017, 1, 1915–1920 RSC.
- M. Haro, C. Sois, G. Molina, L. Otero, J. Bisquert, S. Gimenez and A. Guerrero, Toward Stable Solar Hydrogen Generation Using Organic Photoelectrochemical Cells, J. Phys. Chem. C, 2015, 119, 6488–6494 CrossRef CAS.
- A. Mezzetti, F. Fumagalli, A. Alfano, D. Iadicicco, M. R. Antognazza and F. di Fonzo, Stable Hybrid Organic/Inorganic Photocathodes for Hydrogen Evolution with Amorphous WO3 Hole Selective Contacts, Faraday Discuss., 2017, 198, 433–448 RSC.
- L. Francas, E. Burns, L. Steier, H. Cha, L. Sola-Hernandez, X. O. Li, P. S. Tuladhar, R. Bofill, J. Garcia-Anton and X. Sala, et al., Rational Design of a Neutral Ph Functional and Stable Organic Photocathode, Chem. Commun., 2018, 54, 5732–5735 RSC.
- X. Guo, M. Baumgarten and K. Müllen, Designing Π-Conjugated Polymers for Organic Electronics, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS.
- C. Liu, K. Wang, X. Gong and A. J. Heeger, Low Bandgap Semiconducting Polymers for Polymeric Photovoltaics, Chem. Soc. Rev., 2016, 45, 4825–4846 RSC.
- K. Müllen and W. Pisula, Donor–Acceptor Polymers, J. Am. Chem. Soc., 2015, 137, 9503–9505 CrossRef PubMed.
- K. Kawabata, M. Saito, I. Osaka and K. Takimiya, Very Small Bandgap Π-Conjugated Polymers with Extended Thienoquinoids, J. Am. Chem. Soc., 2016, 138, 7725–7732 CrossRef CAS PubMed.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, For the Bright Future—Bulk Heterojunction Polymer Solar Cells with Power Conversion Efficiency of 7.4%, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed.
- L. Lu and L. Yu, Understanding Low Bandgap Polymer Ptb7 and Optimizing Polymer Solar Cells Based on It, Adv. Mater., 2014, 26, 4413–4430 CrossRef CAS PubMed.
- M. Zhang, X. Guo, S. Zhang and J. Hou, Synergistic Effect of Fluorination on Molecular Energy Level Modulation in Highly Efficient Photovoltaic Polymers, Adv. Mater., 2013, 26, 1118–1123 CrossRef PubMed.
- P. Liu, K. Zhang, F. Liu, Y. Jin, S. Liu, T. P. Russell, H.-L. Yip, F. Huang and Y. Cao, Effect of Fluorine Content in Thienothiophene–Benzodithiophene Copolymers on the Morphology and Performance of Polymer Solar Cells, Chem. Mater., 2014, 26, 3009–3017 CrossRef.
- F. Meyer, Fluorinated Conjugated Polymers in Organic Bulk Heterojunction Photovoltaic Solar Cells, Prog. Polym. Sci., 2015, 47, 70–91 CrossRef CAS.
- S. Nam, J. Seo, S. Woo, W. H. Kim, H. Kim, D. D. C. Bradley and Y. Kim, Inverted Polymer Fullerene Solar Cells Exceeding 10% Efficiency with Poly(2-Ethyl-2-Oxazoline) Nanodots on Electron-Collecting Buffer Layers, Nat. Commun., 2015, 6, 8929 CrossRef CAS PubMed.
- C. Cui, Z. He, Y. Wu, X. Cheng, H. Wu, Y. Li, Y. Cao and W.-Y. Wong, High-Performance Polymer Solar Cells Based on a 2d-Conjugated Polymer with an Alkylthio Side-Chain, Energy Environ. Sci., 2016, 9, 885–891 RSC.
- S. Li, L. Ye, W. Zhao, H. Yan, B. Yang, D. Liu, W. Li, H. Ade and J. Hou, A Wide Band Gap Polymer with a Deep Highest Occupied Molecular Orbital Level Enables 14.2% Efficiency in Polymer Solar Cells, J. Am. Chem. Soc., 2018, 140, 7159–7167 CrossRef CAS PubMed.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Energy-Level Modulation of Small-Molecule Electron Acceptors to Achieve over 12% Efficiency in Polymer Solar Cells, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Aggregation and Morphology Control Enables Multiple Cases of High-Efficiency Polymer Solar Cells, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- J. Zhao, Y. Li, A. Hunt, J. Zhang, H. Yao, Z. Li, J. Zhang, F. Huang, H. Ade and H. Yan, A Difluorobenzoxadiazole Building Block for Efficient Polymer Solar Cells, Adv. Mater., 2015, 28, 1868–1873 CrossRef PubMed.
- J. Zhao, Y. Li, H. Lin, Y. Liu, K. Jiang, C. Mu, T. Ma, J. Y. Lin Lai, H. Hu and D. Yu, et al., High-Efficiency Non-Fullerene Organic Solar Cells Enabled by a Difluorobenzothiadiazole-Based Donor Polymer Combined with a Properly Matched Small Molecule Acceptor, Energy Environ. Sci., 2015, 8, 520–525 RSC.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Efficient Organic Solar Cells Processed from Hydrocarbon Solvents, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma and H. Ade, et al., Fast Charge Separation in a Non-Fullerene Organic Solar Cell with a Small Driving Force, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Röhr, S. Holliday, A. Wadsworth, S. Lockett and M. Neophytou, et al., Reducing the Efficiency–Stability–Cost Gap of Organic Photovoltaics with Highly Efficient and Stable Small Molecule Acceptor Ternary Solar Cells, Nat. Mater., 2016, 16, 363 CrossRef PubMed.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang and N. Gasparini, et al., High-Efficiency and Air-Stable P3HT-Based Polymer Solar Cells with a New Non-Fullerene Acceptor, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- N. Li, J. D. Perea, T. Kassar, M. Richter, T. Heumueller, G. J. Matt, Y. Hou, N. S. Güldal, H. Chen and S. Chen, et al., Abnormal Strong Burn-in Degradation of Highly Efficient Polymer Solar Cells Caused by Spinodal Donor-Acceptor Demixing, Nat. Commun., 2017, 8, 14541 CrossRef CAS PubMed.
- J. Razzell-Hollis, J. Wade, W. C. Tsoi, Y. Soon, J. Durrant and J.-S. Kim, Photochemical Stability of High Efficiency PTB7:PC70BM Solar Cell Blends, J. Mater. Chem. A, 2014, 2, 20189–20195 RSC.
- N. Y. Doumon, G. Wang, R. C. Chiechi and L. J. A. Koster, Relating Polymer Chemical Structure to the Stability of Polymer:Fullerene Solar Cells, J. Mater. Chem. C, 2017, 5, 6611–6619 RSC.
- A. Giovannitti, J. Thorley Karl, B. Nielsen Christian, J. Li, J. Donahue Mary, G. Malliaras George, J. Rivnay and I. McCulloch, Redox-Stability of Alkoxy-Bdt Copolymers and Their Use for Organic Bioelectronic Devices, Adv. Funct. Mater., 2018, 28, 1706325 CrossRef.
- A. J. Pearson, P. E. Hopkinson, E. Couderc, K. Domanski, M. Abdi-Jalebi and N. C. Greenham, Critical Light Instability in CB/DIO Processed PBDTTT-EFT:PC71BM Organic Photovoltaic Devices, Org. Electron., 2016, 30, 225–236 CrossRef CAS.
- B. J. Tremolet de Villers, K. A. O'Hara, D. P. Ostrowski, P. H. Biddle, S. E. Shaheen, M. L. Chabinyc, D. C. Olson and N. Kopidakis, Removal of Residual Diiodooctane Improves Photostability of High-Performance Organic Solar Cell Polymers, Chem. Mater., 2016, 28, 876–884 CrossRef CAS.
- S. Holliday and K. Luscombe Christine, Low Boiling Point Solvent Additives for Improved Photooxidative Stability in Organic Photovoltaics, Adv. Electron. Mater., 2017, 1700416 CrossRef.
- M. O. Reese, M. S. White, G. Rumbles, D. S. Ginley and S. E. Shaheen, Optimal Negative Electrodes for Poly(3-Hexylthiophene): [6,6]-Phenyl C61-Butyric Acid Methyl Ester Bulk Heterojunction Photovoltaic Devices, Appl. Phys. Lett., 2008, 92, 053307 CrossRef.
- B. Xu, S.-A. Gopalan, A.-I. Gopalan, N. Muthuchamy, K.-P. Lee, J.-S. Lee, Y. Jiang, S.-W. Lee, S.-W. Kim and J.-S. Kim, et al., Functional Solid Additive Modified PEDOT:Pss as an Anode Buffer Layer for Enhanced Photovoltaic Performance and Stability in Polymer Solar Cells, Sci. Rep., 2017, 7, 45079 CrossRef CAS PubMed.
- C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Non-Fullerene Electron Acceptors for Use in Organic Solar Cells, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef CAS PubMed.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Next-Generation Organic Photovoltaics Based on Non-Fullerene Acceptors, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Critical Review of the Molecular Design Progress in Non-Fullerene Electron Acceptors Towards Commercially Viable Organic Solar Cells, Chemical Society Reviews, 2018, 10.1039/c7cs00892a.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Nonfullerene Acceptor Molecules for Bulk Heterojunction Organic Solar Cells, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS PubMed.
- S. Holliday, R. S. Ashraf, C. B. Nielsen, M. Kirkus, J. A. Röhr, C.-H. Tan, E. Collado-Fregoso, A.-C. Knall, J. R. Durrant and J. Nelson, et al., A Rhodanine Flanked Nonfullerene Acceptor for Solution-Processed Organic Photovoltaics, J. Am. Chem. Soc., 2015, 137, 898–904 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, An Electron Acceptor Challenging Fullerenes for Efficient Polymer Solar Cells, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Fullerene-Free Polymer Solar Cells with over 11% Efficiency and Excellent Thermal Stability, Adv. Mater., 2016, 28, 4734–4739 CrossRef CAS PubMed.
- H.-H. Gao, Y. Sun, X. Wan, X. Ke, H. Feng, B. Kan, Y. Wang, Y. Zhang, C. Li and Y. Chen, A New Nonfullerene Acceptor with near Infrared Absorption for High Performance Ternary-Blend Organic Solar Cells with Efficiency over 13%, Adv. Sci., 2018, 5, 1800307 CrossRef PubMed.
- H. Yao, Y. Cui, R. Yu, B. Gao, H. Zhang and J. Hou, Design, Synthesis, and Photovoltaic Characterization of a Small Molecular Acceptor with an Ultra-Narrow Band Gap, Angew. Chem., Int. Ed., 2017, 56, 3045–3049 CrossRef CAS PubMed.
- D. Baran, T. Kirchartz, S. Wheeler, S. Dimitrov, M. Abdelsamie, J. Gorman, R. S. Ashraf, S. Holliday, A. Wadsworth and N. Gasparini, et al., Reduced Voltage Losses Yield 10% Efficient Fullerene Free Organic Solar Cells with >1 V Open Circuit Voltages, Energy Environ. Sci., 2016, 9, 3783–3793 RSC.
- H.-Y. Chen, J. Golder, S.-C. Yeh, C.-W. Lin, C.-T. Chen and C.-T. Chen, Diindeno[1,2-G:1′,2′-S]Rubicene: All-Carbon Non-Fullerene Electron Acceptor for Efficient Bulk-Heterojunction Organic Solar Cells with High Open-Circuit Voltage, RSC Adv., 2015, 5, 3381–3385 RSC.
- H. Cha, J. Wu, A. Wadsworth, J. Nagitta, S. Limbu, S. Pont, Z. Li, J. Searle, F. Wyatt Mark and D. Baran, et al., An Efficient, “Burn in” Free Organic Solar Cell Employing a Nonfullerene Electron Acceptor, Adv. Mater., 2017, 29, 1701156 CrossRef PubMed.
- X. Yang, J. K. J. van Duren, R. A. J. Janssen, M. A. J. Michels and J. Loos, Morphology and Thermal Stability of the Active Layer in Poly(P-Phenylenevinylene)/Methanofullerene Plastic Photovoltaic Devices, Macromolecules, 2004, 37, 2151–2158 CrossRef CAS.
- X. Yang, J. Loos, S. C. Veenstra, W. J. H. Verhees, M. M. Wienk, J. M. Kroon, M. A. J. Michels and R. A. J. Janssen, Nanoscale Morphology of High-Performance Polymer Solar Cells, Nano Lett., 2005, 5, 579–583 CrossRef CAS PubMed.
- C. Schroeder Bob, Z. Li, A. Brady Michael, C. Faria Gregório, S. Ashraf Raja, J. Takacs Christopher, S. Cowart John, T. Duong Duc, H. Chiu Kar and C.-H. Tan, et al., Enhancing Fullerene-Based Solar Cell Lifetimes by Addition of a Fullerene Dumbbell, Angew. Chem., Int. Ed., 2014, 53, 12870–12875 CrossRef CAS PubMed.
- V. I. Madogni, B. Kounouhéwa, A. Akpo, M. Agbomahéna, S. A. Hounkpatin and C. N. Awanou, Comparison of Degradation Mechanisms in Organic Photovoltaic Devices Upon Exposure to a Temperate and a Subequatorial Climate, Chem. Phys. Lett., 2015, 640, 201–214 CrossRef CAS.
- M. Wang and I. G. Hill, Fluorinated Alkyl Phosphonic Acid Sams Replace PEDOT:Pss in Polymer Semiconductor Devices, Org. Electron., 2012, 13, 498–505 CrossRef CAS.
- L.-M. Chen, Z. Hong, G. Li and Y. Yang, Recent Progress in Polymer Solar Cells: Manipulation of Polymer:Fullerene Morphology and the Formation of Efficient Inverted Polymer Solar Cells, Adv. Mater., 2009, 21, 1434–1449 CrossRef CAS.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Enhanced Power-Conversion Efficiency in Polymer Solar Cells Using an Inverted Device Structure, Nat. Photonics, 2012, 6, 591 CrossRef.
- Z. Xu, L.-M. Chen, G. Yang, C.-H. Huang, J. Hou, Y. Wu, G. Li, C.-S. Hsu and Y. Yang, Vertical Phase Separation in Poly(3-Hexylthiophene): Fullerene Derivative Blends and Its Advantage for Inverted Structure Solar Cells, Adv. Funct. Mater., 2009, 19, 1227–1234 CrossRef CAS.
- Y. Yan, X. Liu and T. Wang, Conjugated-Polymer Blends for Organic Photovoltaics: Rational Control of Vertical Stratification for High Performance, Adv. Mater., 2017, 29, 1601674 CrossRef PubMed.
- T. Ameri, G. Dennler, C. Lungenschmied and C. J. Brabec, Organic Tandem Solar Cells: A Review, Energy Environ. Sci., 2009, 2, 347–363 RSC.
- T. Ameri, N. Li and C. J. Brabec, Highly Efficient Organic Tandem Solar Cells: A Follow up Review, Energy Environ. Sci., 2013, 6, 2390–2413 RSC.
- G. Li, W.-H. Chang and Y. Yang, Low-Bandgap Conjugated Polymers Enabling Solution-Processable Tandem Solar Cells, Nat. Rev. Mater., 2017, 2, 17043 CrossRef CAS.
- J. T. Kirner and R. G. Finke, Water-Oxidation Photoanodes Using Organic Light-Harvesting Materials: A Review, J. Mater. Chem. A, 2017, 5, 19560–19592 RSC.
- J. Zou, C.-Z. Li, C.-Y. Chang, H.-L. Yip and K. Y. Jen Alex, Interfacial Engineering of Ultrathin Metal Film Transparent Electrode for Flexible Organic Photovoltaic Cells, Adv. Mater., 2014, 26, 3618–3623 CrossRef CAS PubMed.
- X. Che, Y. Li, Y. Qu and S. R. Forrest, High Fabrication Yield Organic Tandem Photovoltaics Combining Vacuum- and Solution-Processed Subcells with 15% Efficiency, Nat. Energy, 2018, 3, 422–427 CrossRef CAS.
- Y. Gao, M. Le Corre Vincent, A. Gaïtis, M. Neophytou, A. Hamid Mahmoud, K. Takanabe and M. Beaujuge Pierre, Homo-Tandem Polymer Solar Cells with VOC > 1.8 V for Efficient Pv-Driven Water Splitting, Adv. Mater., 2016, 28, 3366–3373 CrossRef CAS PubMed.
- J. You, C.-C. Chen, Z. Hong, K. Yoshimura, K. Ohya, R. Xu, S. Ye, J. Gao, G. Li and Y. Yang, 10.2% Power Conversion Efficiency Polymer Tandem Solar Cells Consisting of Two Identical Sub-Cells, Adv. Mater., 2013, 25, 3973–3978 CrossRef CAS PubMed.
- S. Esiner, H. van Eersel, M. Wienk Martijn and A. J. Janssen René, Triple Junction Polymer Solar Cells for Photoelectrochemical Water Splitting, Adv. Mater., 2013, 25, 2932–2936 CrossRef CAS PubMed.
- S. Esiner, G. W. P. van Pruissen, M. M. Wienk and R. A. J. Janssen, Optimized Light-Driven Electrochemical Water Splitting with Tandem Polymer Solar Cells, J. Mater. Chem. A, 2016, 4, 5107–5114 RSC.
- S. Esiner, R. E. M. Willems, A. Furlan, W. Li, M. M. Wienk and R. A. J. Janssen, Photoelectrochemical Water Splitting in an Organic Artificial Leaf, J. Mater. Chem. A, 2015, 3, 23936–23945 RSC.
- S. Chen, G. Zhang, J. Liu, H. Yao, J. Zhang, T. Ma, Z. Li and H. Yan, An All-Solution Processed Recombination Layer with Mild Post-Treatment Enabling Efficient Homo-Tandem Non-Fullerene Organic Solar Cells, Adv. Mater., 2016, 29, 1604231 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |