
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silicon monophosphide as a possible lithium battery anode material†
R.
Reinhold
 *ab,
D.
Mikhailova
a,
T.
Gemming
a,
A. B.
Missyul
c,
C.
Nowka
a,
S.
Kaskel
b and
L.
Giebeler
*a
*ab,
D.
Mikhailova
a,
T.
Gemming
a,
A. B.
Missyul
c,
C.
Nowka
a,
S.
Kaskel
b and
L.
Giebeler
*a
aLeibniz Institute for Solid State and Materials Research (IFW) Dresden e.V., Helmholtzstraße 20, D-01069 Dresden, Germany. E-mail: l.giebeler@ifw-dresden.de; r.reinhold@ifw-dresden.de
bDepartment of Inorganic Chemistry, Technische Universität Dresden, Bergstraße 66, D-01069 Dresden, Germany
cCELLS-ALBA Synchrotron, Carrer de la Llum 2-26, 08290 Cerdanyoladel Vallèes, Barcelona, Spain
First published on 12th October 2018
Abstract
Binary (semi)metal–phosphorus compounds fascinate nowadays as they promise high specific capacities in metal ion batteries and an advantageous in situ formation of the electrochemically active material inside the electrochemical cell without air exposure. Here, we report on SiP as a new member of this material family which was tested as an anode material in Li ion batteries due to a highly attractive theoretical specific capacity of about 3000 mAh g−1 SiP. After synthesis by a vapour-transport reaction a cotton wool-like product was obtained revealing a layered 2D crystalline microribbon-like morphology, space group Cmc21, which may allow fast Li ion intercalation/diffusion kinetics. SiP also opens up a novel perspective for application in ultra-thin optoelectronics or flexible photovoltaics. During the first discharging half-cycle the crystalline phase amorphizes as indicated by operando synchrotron powder diffraction followed by an amorphous state without re-formation of any crystalline phase, including Li3P. Our investigations after 50 discharging–charging cycles show a specific capacity of 550 mAh g−1, which supports the trend of lifetime reduction by LiP formation. Further optimisation is necessary to allow a broad application for novel technologies like high performance Li ion or Li–S batteries.
Lithium ion batteries have revolutionized the mobile devices market starting in the early 1990s and are just about to do the same in the automotive and even in the stationary storage sectors. To allow for higher efficiency batteries with much increased electrical energy storage capacities, many efforts have focused on introducing high power, high performance materials, e.g. to achieve customers' range demands for e-vehicles. Here, the classical graphite negative electrode reaches its limit when much higher lithium uptake is necessary as typical for Li–S batteries. Silicon compounds represent highly promising candidates to replace graphite as the anode material as already partially realized in commercial battery technology.1–4 However, volume expansion and consequently pulverization5 of the material is still the greatest obstacle to solve which, in consequence, leads to conductivity losses in the electrode composite and the repeated formation of new surfaces supporting parasitic electrolyte consumption.6 Many studies therefore focused on the design of nano-silicon morphologies such as nanowires,7–10 nanoparticles11,12 or nanopillars.13,14 Nanoparticles below 20 nm also serve as an easy-to-reach alternative to the high-tech preparation of nanoarchitectures and show similar performance in metal ion batteries. An intelligent gimmick in the sense of nanoparticle synthesis is the application of transition metal phosphides (MP, M = Cu, Co, Sn).15–19 By a complete conversion Li3P and active metallic nanoparticles are formed in situ.
In particular, studies regarding Sn4P3 show a broad applicability in lithium,17,20 sodium21–23 and potassium24,25 ion batteries. Due to the similar chemical properties, the electrochemical behaviour of binary Si–P compounds should be similar. However, this work focuses on lithium-based batteries as silicon shows a high ability to form intermetallic phases with lithium compared to sodium or potassium. To reach high specific capacities low elemental weights are recommended and can be realized by combining silicon and phosphorus. Si–P-compounds represent a new class of future anode materials where silicon diphosphide (SiP2) has already attracted high attention due to its high theoretical capacity of 2900 mAh g−1 (Li15Si4, Li3P).26–28
The high abundance of silicon and phosphorus combined with their electrochemical mechanism of alloying and conversion mechanism justifies a scientific effort to implement the material into metal ion batteries. A relatively fast deactivation of SiP2 by successive and irreversible LiP shell formation establishes an insulating layer around the Si particle core.28 Reducing the phosphorus content, as in the case of silicon monophosphide, benefits an even higher theoretical specific capacity (3060 mAh g−1; Li15Si4, Li3P) compared to SiP2 and may avoid excessive LiP formation. Moreover, SiP is expected to form in situ electrochemically active silicon nanoparticles at reduced volume changes. Addressing the latter two scenarios simultaneously, the lifetime of the battery may be significantly increased, especially compared to SiP2.28 An interesting feature of the used synthesis is found in the self-organization of SiP in a layered 2D ribbon-like material in a bottom-up approach without additional handling and forms Si nanoparticles in situ during the electrochemical reaction, e.g. without further temperature treatment and no complex deposition methods are applied as needed for other nano-sized silicon morphologies.7–10,13,14
From the Si–P binary phase diagram developed by Olesinski,29 silicon monophosphide is the only stable phase under ambient pressure conditions, emphasizing its thermodynamic stability compared to SiP2. Regarding silicon monophosphide, only a few synthesis procedures have been described in combination with crystal structure studies. First syntheses were realized in the 1960s via vapour-growth reactions30,31 and recently by a high pressure-high temperature synthesis in a cubic multi-anvil cell32 leading to bulk crystal materials for both techniques. This group IV–V monopnictide SiP shows an orthorhombic (Cmc21 space group) 2D layered structure, where single layers are stacked by weak van der Waals interactions.33 First DFT calculations33 demonstrated comparable exfoliation energies as for graphite which enable exfoliation by mechanical stress resulting in semiconducting SiP layers as an interesting candidate for applications in thin, bendable photovoltaics and optoelectronics.33
We report on the optimized SiP synthesis via a chemical vapour-transport reaction obtaining layered 2D materials and give for the first time deeper insights into the structural properties and electrochemical behaviour. This work mainly focuses on the electrochemical properties of SiP as a negative electrode material for lithium ion batteries. Another key point considers the structural changes during lithiation to get an idea on a possible structure preservation which may be reachable with the observed layered arrangement as would be untypical for phosphide compounds when treated under similar electrochemical conditions where they undergo a conversion reaction to the elemental state and usually Li3P.
Our results reveal moderate electrochemical activity and are still under improvement to enhance the specific capacity and lifetime.
After synthesis with an exothermic chemical vapour-transport reaction (T1→T2, T2 > T1), a cotton wool-like product was obtained at T2 = 1000 °C revealing a layered 2D microribbon-shaped morphology as photographed by SEM (Fig. 1) where the macroscopically visible, exfoliatable layers are marked with arrows in the inset. The corresponding EDXS mappings, displayed in Fig. S-1,† confirm the homogeneous distribution of Si and P throughout the sample.
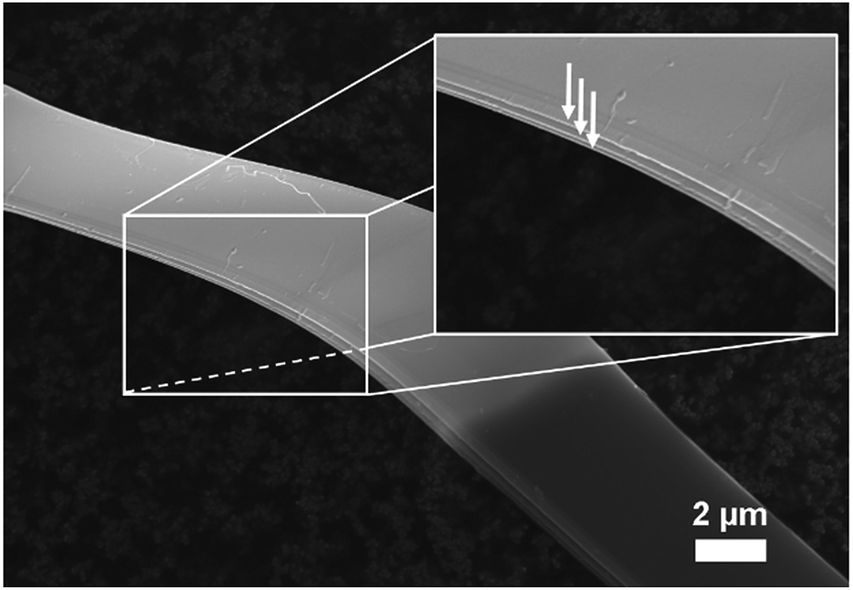 | ||
| Fig. 1 SEM image of as-prepared SiP. Macroscopic exfoliatable layers are indicated by arrows in the inset. | ||
The powder XRD pattern of the as-synthesized SiP (Fig. 2) is in good agreement with literature data30,31 and is analysed with the corresponding SiP structure model with orthorhombic space group Cmc21 as shown in Fig. S-2.† SiP lattice parameters were determined by a Le Bail fit to a = 3.51473(7) Å, b = 20.4880(5) Å, c = 13.6198(4) Å. The structure is described as layered in the (001) direction. Due to the weak van der Waals bonds between the layers, SiP may be favourable for ion intercalation as well as exfoliation of individual layers. Selected area electron diffraction (SAED) (Fig. 3) shows that a ribbon of as-prepared SiP is characterized by a single crystalline habit due to the absence of additional, extrinsic diffraction spots or diversely oriented individuals like typical for twins.31
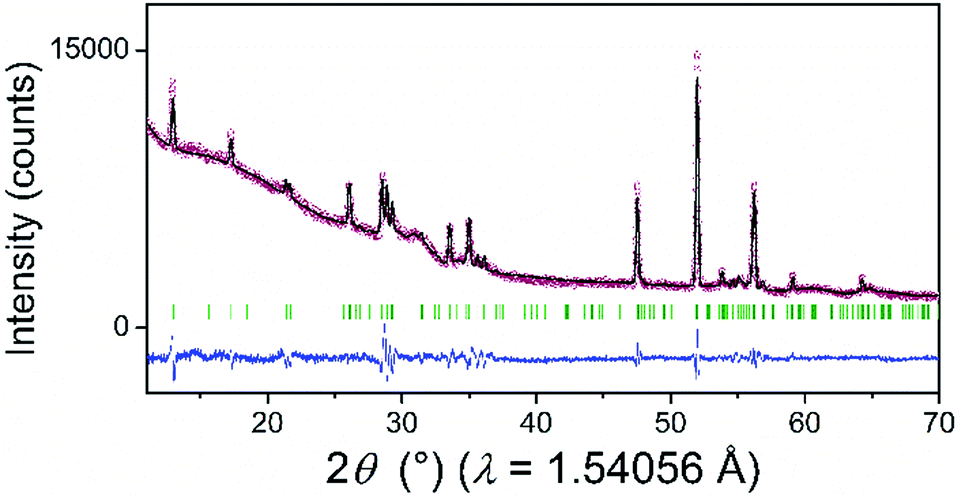 | ||
| Fig. 2 XRD pattern of the as-prepared SiP (red dots) with the calculated profile (black solid line) according to the Le Bail analysis of the structural model from SiP. The difference curve resulted from the subtraction of the observed from the calculated data (blue solid line). Green vertical lines represent the Bragg positions of the reflections according to the SiP–space group Cmc21–structural model known from the literature.30,31 | ||
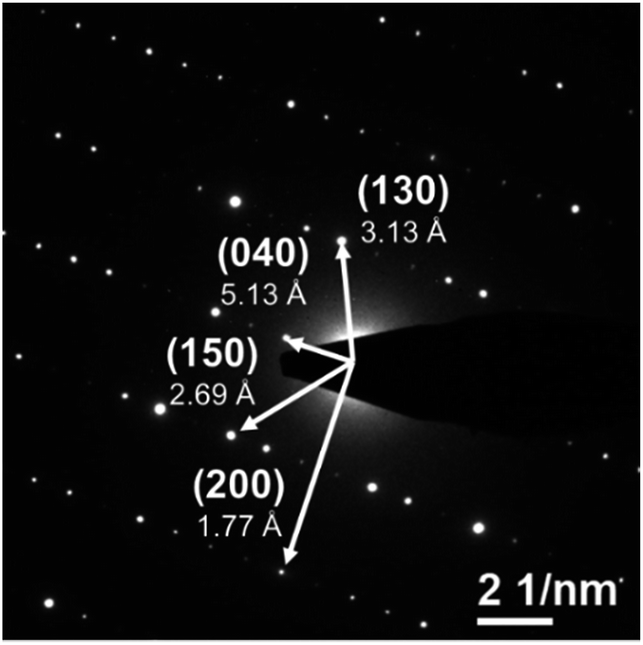 | ||
| Fig. 3 SAED pattern of as-prepared SiP with indexed diffraction spots along the (001) zone axis. | ||
Additionally, high resolution transmission electron microscopy (HRTEM) images (Fig. S-3 and S-4†) nicely present the homogeneous arrangement of atoms within the layers. STEM-EDXS measurements (Fig. S-5†) were recorded to verify the elemental composition. The spectra perfectly support the expected content of Si-to-P ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and show no signal for iodine (transporting agent). As previously reported,28 SiP2 reacts during lithiation in a three-step mechanism: (1) a conversion reaction to intermediate LiPx phases (1 ≤ x ≤ 7) followed by (2) the formation of Li3P, and, finally, (3) the alloying of Li and Si to LixSi. In comparison to SiP2, the differential capacity plot (DCP) of SiP during the first cycle, displayed in Fig. 4, shows similar redox behaviour. For further discussion, discharging is defined as insertion of Li ions while charging describes the removal. For better illustration, the first derivative of the current is calculated normalized to the mass of SiP.
:1 and show no signal for iodine (transporting agent). As previously reported,28 SiP2 reacts during lithiation in a three-step mechanism: (1) a conversion reaction to intermediate LiPx phases (1 ≤ x ≤ 7) followed by (2) the formation of Li3P, and, finally, (3) the alloying of Li and Si to LixSi. In comparison to SiP2, the differential capacity plot (DCP) of SiP during the first cycle, displayed in Fig. 4, shows similar redox behaviour. For further discussion, discharging is defined as insertion of Li ions while charging describes the removal. For better illustration, the first derivative of the current is calculated normalized to the mass of SiP.
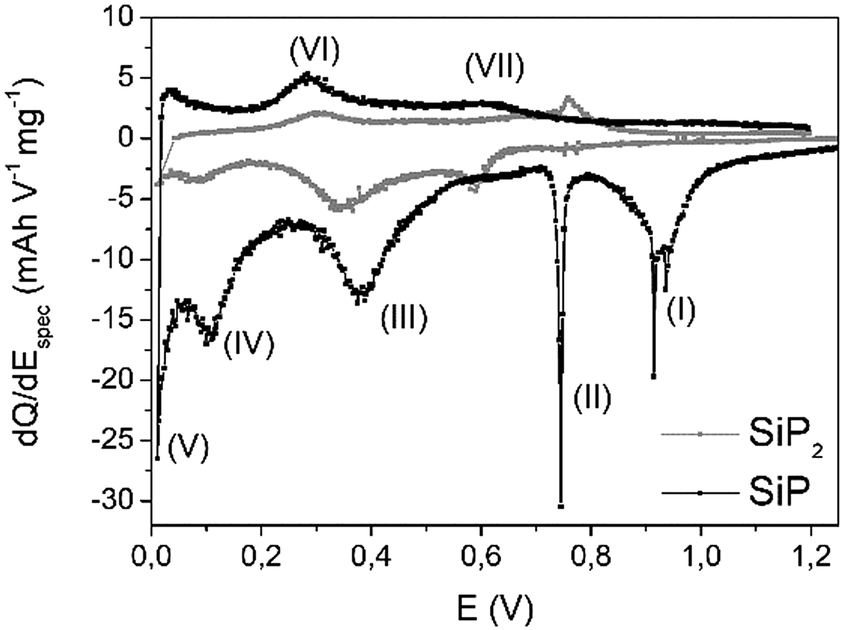 | ||
| Fig. 4 Differential capacity plot (DCP) of SiP (black) during the first cycle. For comparison, DCP of SiP2 (grey) is given as a reference. | ||
Comparing the potential window of 0.8–1.0 V vs. Li/Li+, various redox peaks (I+II) occur, which most likely indicate structural changes due to the sharp signals. This is closely associated with a relatively low kinetic barrier. Based on the absence of both signals (Fig. S-6†) in the second cycle, irreversible processes like the formation of a solid electrolyte interphase (SEI) or the lithiation of a carbon additive at 0.74 V might be possible.34
The reaction to Li3P (III) proceeds around 0.4 V and nicely fits the previous results of SiP2.28 Below 0.1 V, alloying of silicon nanoparticles (IV, V) should occur. During delithiation, reaction (IV) at 320 mV may correspond to the dealloying of LixSi particles, which is also observed for pure silicon in the same range.11,35 At about 0.6 V vs. Li/Li+ during Li removal, the decomposition of Li3P species proceeds. Interestingly, all lithiation processes are slightly shifted to higher voltages whereas the corresponding delithiation reactions are found at somewhat lower voltages leading to the assumption of faster kinetics. Comparing the currents of both redox processes, a higher current is observed during the lithiation process implying a low coulombic efficiency in the first cycle.
For further insights on possible changes, e.g. structural transitions, phase formations, dependent on the scanning voltage, operando synchrotron XRD was performed and the results are displayed in Fig. 5.
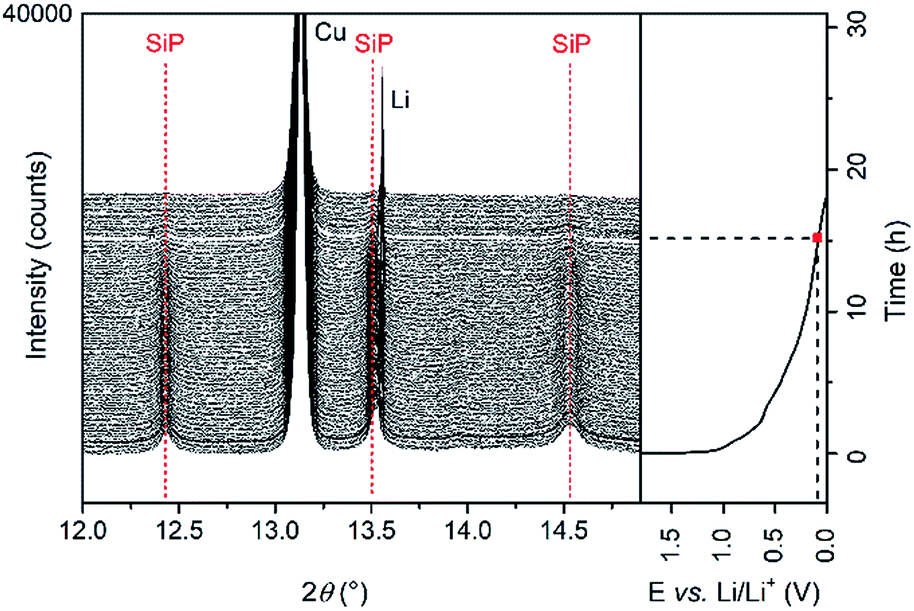 | ||
| Fig. 5 Operando synchrotron XRD pattern collected during the first discharging cycle. Red dashed lines indicate vanishing reflections. | ||
For better clarity, only the diffraction patterns between 12° and 15° 2θ are presented. The intensity of the reflections of the crystalline SiP (indexed with red dashed lines) decreases continuously and vanishes at voltages below 0.1 V vs. Li/Li+. During Li insertion into the active material, no shift of the reflections to higher or lower angles is observed. According to these results, an intercalation of Li ions in-between the SiP layers like for graphite is excluded. Moreover, in the fully lithiated state, no reflections of crystalline Li3P appear, which is in contrast to our previous study regarding SiP2. Three reasons can explain this behaviour. (1) The Li intercalation mechanism in the case of SiP differs from the one of SiP2. (2) Li3P is formed but the critical size of the particles to induce crystallization has not been reached in the first discharging cycle. Li3P therewith may exist in an amorphous state. (3) Phosphorus only reacts to LiP which, however, is amorphous as reported previously for SiP2.28 For further analysis of the electrochemical performance, galvanostatic cycling of SiP electrodes in a two-electrode configuration vs. Li/Li+ was conducted. The SiP cells were cycled with a current density of 100 mA g−1, which corresponds to a C-rate of about C/30.
In the first charging cycle, capacities of 1000 mAh g−1 (Fig. 6) are retained which highlights the potential of this anode material. As expected from DCP during the first cycle, low coulombic efficiencies of 32% are observed. This observation is most likely attributed to structural changes and irreversible reactions, SEI formation, etc. during cycling. The formation of an insulating LiP phase as observed for other metal phosphides might be possible but we have not found any evidence for it. Further investigations regarding intermediate phases are in progress. However, after 50 cycles, delithiation capacity remains constant at around 550 mAh g−1 showing high desirable coulombic efficiencies of 99.6%. Additionally, SiP offers a good rate capability at high current densities of up to 500 mA g−1, as displayed in Fig. S-7.†
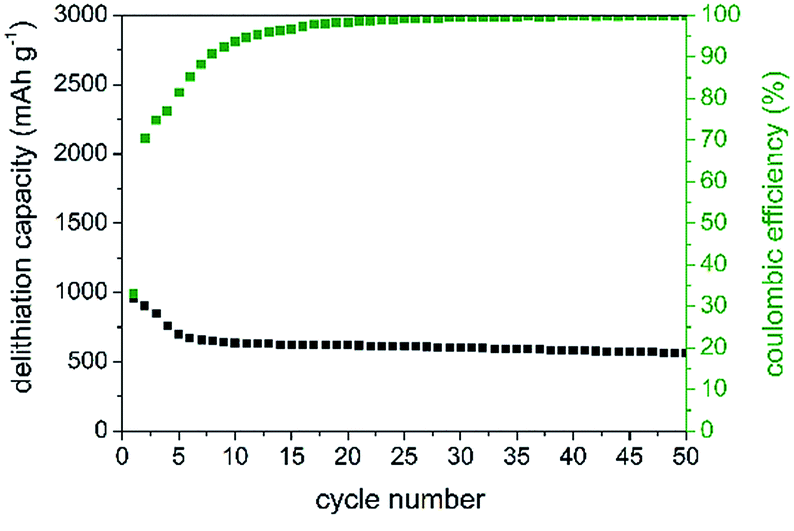 | ||
| Fig. 6 Delithiation capacity and corresponding coulombic efficiencies (green) of SiP. | ||
Conclusions
We herein reported an optimized synthesis of SiP via a vapour-transport reaction using iodine as the transport agent. As a product, microribbon-like SiP with an orthorhombic Cmc21 space group is obtained. TEM measurements combined with XRD confirm the crystal structure and elemental ratio of 1:1 (Si:P). First electrochemical studies highlighted its high potential as an anode material since a capacity of 1000 mAh g−1 after the first discharging cycle was demonstrated. However, the material suffers from fast capacity fading due to an obviously incomplete reaction. Therefore, further conceptual development is required to avoid the LiP insulating shell formation.
Experimental section
Synthesis of SiP
Silicon monophosphide (SiP) was synthesized via a transport reaction of pure silicon (Aldrich, −325 mesh, 99% purity) and red phosphorus (Alfa Aesar, −100 mesh, 98.9% purity) powders. Iodine (Merck, 99%) was added as a transporting agent. 50 mg of a stoichiometric mixture of silicon and phosphorus and 6 mg of iodine (placed in an extra tube) were filled in a quartz ampoule. The ampoule was sealed under vacuum (10−3 mbar) and was placed into a horizontal two-zone furnace. In a first equilibrating step, the temperature was set to 400 °C for 16 h to guarantee an equilibrium state of phosphorus partial pressure. In a second step, the temperature was increased to 900 °C (T1, source) and 1000 °C (T2, sink) and kept for 160 h to allow the exothermic reaction to proceed. This temperature gradient was chosen based on the studies on other metalphosphides.36 Finally, the ampoule was cooled down naturally. All following steps to work with the SiP were conducted under an inert gas atmosphere in a glovebox.Material characterization
X-ray diffraction (XRD) patterns were recorded on a Stadi P diffractometer (STOE) with a curved Ge(111)-crystal as the monochromator. Samples were filled in a glass capillary (Hilgenberg, Nr. 10, diameter 0.7 mm) and fire-sealed. Cu Kα1 radiation (λ = 1.54056 Å) was applied. Capillaries were measured in Debye–Scherrer mode using a step width of Δ2θ = 0.02°. FullProf implemented into the software WinPlotR was used for performing Le Bail analyses. The patterns were analysed according to literature data for SiP.30Operando powder synchrotron diffraction measurements on SiP in a coin cell with a glass window of 3 mm in diameter were performed at the beamline BL04-MSPD37 at ALBA (Barcelona, Spain) in a transmission mode. A Li disk served as an anode. The experimental setup containing a coin cell holder connected to a VMP multichannel potentiostat is described elsewhere.38 Data were collected every 9 minutes in steps of 0.005° at λ = 0.41310(1) Å, which were refined from the reflection positions of a NIST LaB6 reference material.
In order to characterize pristine SiP, a pattern was recorded before starting the electrochemical process. The cell was then successively discharged in galvanostatic mode at a constant current of 100 mA g−1. The Cu mesh current collector on the positive electrode side was used as an internal standard during the measurements.
Scanning electron microscopy (SEM) was performed on a Gemini LEO 1530 (Zeiss) with an acceleration voltage of 10 kV.
Transmission electron microscopy (TEM) experiments were carried out on a FEI Tecnai F30 with 300 kV acceleration voltage equipped with a field emission gun. The material was dispersed in dimethyl carbonate (DMC) through sonication and drop coated onto a copper grid with a lacey carbon layer as the sample holder. Selected area electron diffraction (SAED), high resolution transmission electron microscopy (HRTEM) images and scanning transmission electron microscopy energy-dispersive X-ray spectroscopy (STEM-EDX) images were taken.
Electrode preparation
For electrode preparation, 30 wt% SiP was mixed with 60 wt% Super P (Timcal) and 10 wt% PVDF 1013 (Solvay). The mixture was dissolved in N-methylpyrrolidone (NMP, Sigma Aldrich), drop-coated onto a copper foil (∅ 12 mm) and dried at 80 °C overnight under vacuum. For assembling the cells in a two-electrode configuration, a lithium metal disc (Chempur, 250 μm thickness), two glass fibre separators (Whatman) and 250 μl electrolyte (1 M LiPF6 in dimethyl carbonate (DMC)/ethylene carbonate (EC) (1:1 v/v), LP30, BASF) and the SiP composite as the working electrode were arranged in a Swagelok cell casing. Electrochemical tests were realized with a multichannel VMP3 potentiostat (BioLogic) at a constant temperature of 25 °C. Galvanostatic cycling with potential limitation (GCPL) was conducted between 0.01 and 1.2 V vs. Li/Li+ at a current density of 100 mA g−1. For further characterization, different current densities in the range of 50–200 mA g−1 were applied. The specific capacity and current density were calculated based on the mass of silicon monophosphide.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank K. Wruck for assisting us in the synthesis of SiP. Beamtime allocation at the BL04-MSPD beamline at the ALBA Synchrotron in Barcelona, Spain, is gratefully acknowledged. This work was partially supported with funding of the German Federal Ministry of Education and Research (BMBF) in the WING center BamoSa– Battery mobility in Saxony (grant no. 03X4637) and the European Union/European Regional Development Fund (ERDF) and the Free State of Saxony in the TTKin project (SAB grant no. 100225299).Notes and references
- M. Jeong, H. L. Du, M. Islam, J. K. Lee, Y. Sun and H. Jung, Nano Lett., 2017, 17, 5600–5606 CrossRef CAS PubMed.
- I. H. Son, J. H. Park, S. Kwon, S. Park, M. H. Rümmeli, A. Bachmatiuk, H. J. Song, J. Ku, J. W. Choi, J. Choi, S.-G. Doo and H. Chang, Nat. Commun., 2015, 6, 7393 CrossRef CAS PubMed.
- D. Nan, Z. Huang, R. Lv, Y. Lin, L. Yang, X. Yu, L. Ye, W. Shen, H. Sun and F. Kang, J. Nanomater., 2014, 139639 Search PubMed.
- M. N. Obrovac and L. J. Krause, J. Electrochem. Soc., 2007, 154, A103–A108 CrossRef CAS.
- M. N. Obrovac and L. Christensen, Electrochem. Solid-State Lett., 2004, 7, A93–A96 CrossRef CAS.
- T. Jaumann, J. Balach, U. Langklotz, V. Sauchuk, M. Fritsch, A. Michaelis, V. Teltevskij, D. Mikhailova, S. Oswald, M. Klose, G. Stephani, R. Hauser, J. Eckert and L. Giebeler, Energy Storage Materials, 2017, 6, 26–35 CrossRef.
- L.-F. Cui, R. Ruffo, C. K. Chan, H. Peng and Y. Cui, Nano Lett., 2009, 9, 491–495 CrossRef CAS PubMed.
- B. Laïk, L. Eude, J. P. Pereira-Ramos, C. S. Cojocaru, D. Pribat and E. Rouvière, Electrochim. Acta, 2008, 53, 5528–5532 CrossRef.
- A. Krause, S. Dörfler, M. Piwko, F. M. Wisser, T. Jaumann, E. Ahrens, L. Giebeler, H. Althues, S. Schädlich, J. Grothe, A. Jeffery, M. Grube, J. Brückner, J. Martin, J. Eckert, S. Kaskel, T. Mikolajick and W. M. Weber, Sci. Rep., 2016, 6, 27982 CrossRef CAS PubMed.
- M. Green and F.-M. Liu, A method of fabricating fibres composed of silicon or a silicon-based material and their use in lithium rechargeable batteries, WO 2007/083155 A1, 2007, pp. 1–14.
- T. Jaumann, M. Herklotz, M. Klose, K. Pinkert, S. Oswald, J. Eckert and L. Giebeler, Chem. Mater., 2015, 27, 37–43 CrossRef CAS.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522–1531 CrossRef CAS PubMed.
- S. W. Lee, M. T. McDowell, J. W. Choi and Y. Cui, Nano Lett., 2011, 11, 3034–3039 CrossRef CAS PubMed.
- M. Green, E. Fielder, B. Scrosati, M. Wachtler and J. S. Moreno, Electrochem. Solid-State Lett., 2003, 6, A75–A79 CrossRef CAS.
- V. Pralong, D. C. S. Souza, K. T. Leung and L. F. Nazar, Electrochem. Commun., 2002, 4, 516–520 CrossRef CAS.
- R. Alcántara, J. L. Tirado, J. C. Jumas, L. Monconduit and J. Olivier-Fourcade, J. Power Sources, 2002, 109, 308–312 CrossRef.
- Y. Kim, C. K. Lee, H. Sohn and T. Kang, J. Electrochem.Soc., 2004, 151, A933–A937 CrossRef CAS.
- B. Mauvernay, M. L. Doublet and L. Monconduit, J. Phys. Chem. Solids, 2006, 67, 1252–1257 CrossRef CAS.
- O. Crosnier and L. F. Nazar, Electrochem. Solid-State Lett., 2004, 7, A187–A189 CrossRef CAS.
- J. Wu and Z. Fu, J. Electrochem. Soc., 2009, 156, A22–A26 CrossRef CAS.
- J. Liu, P. Kopold, C. Wu, P. A. van Aken, J. Maier and Y. Yu, Energy Environ. Sci., 2015, 8, 3531–3538 RSC.
- J. Qian, Y. Xiong, Y. Cao, X. Ai and H. Yang, Nano Lett., 2014, 14, 1865–1869 CrossRef CAS PubMed.
- J. Mao, X. Fan, C. Luo and C. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 7147–7155 CrossRef CAS PubMed.
- W. Zhang, J. Mao, S. Li, Z. Chen and Z. Guo, J. Am. Chem. Soc., 2017, 139, 3316–3319 CrossRef CAS PubMed.
- W. Zhang, W. K. Pang, V. Sencadas and Z. Guo, Joule, 2018, 2, 1534–1547 CrossRef CAS.
- D. Duveau, S. Sananes Israel, J. Fullenwarth, F. Cunin and L. Monconduit, J. Mater. Chem. A, 2016, 4, 3228–3232 RSC.
- H. Kwon, C. K. Lee, K. Jeon and C. Park, ACS Nano, 2016, 10, 5701–5709 CrossRef CAS PubMed.
- R. Reinhold, U. Stoeck, H.-J. Grafe, D. Mikhailova, T. Jaumann, S. Oswald, S. Kaskel and L. Giebeler, ACS Appl. Mater. Interfaces, 2018, 10, 7096–7106 CrossRef CAS PubMed.
- R. W. Olesinski, N. Kanani and G. J. Abbaschian, Bull. Alloy Phase Diagrams, 1985, 6, 130–133 CrossRef CAS.
- T. Wadsten, Acta Chem. Scand., 1969, 23, 2532–2560 CrossRef CAS.
- C. G. Beck and R. Stickler, J. Appl. Phys., 1966, 37, 4683–4687 CrossRef CAS.
- C. Barreteau, B. Michon, C. Besnard and E. Giannini, J. Cryst. Growth, 2016, 443, 75–80 CrossRef CAS.
- A.-Q. Cheng, Z. He, J. Zhao, H. Zeng and R.-S. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 5133–5139 CrossRef CAS PubMed.
- B. Peng, Y. Xu, X. Wang, X. Shi and F. M. Mulder, Sci. China: Phys., Mech. Astron., 2017, 60, 064611 Search PubMed.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966–4985 CrossRef CAS PubMed.
- R. Glaum and R. Gruehn, Z. Anorg. Allg. Chem., 1989, 573, 24–42 CrossRef CAS.
- F. Fauth, I. Peral, C. Popescu and M. Knapp, Powder Diffr., 2013, 28, S360–S370 CrossRef CAS.
- M. Herklotz, J. Weiß, E. Ahrens, M. Yavuz, L. Mereacre, N. Kiziltas-Yavuz, C. Dräger, H. Ehrenberg, J. Eckert, F. Fauth, L. Giebeler and M. Knapp, J. Appl. Crystallogr., 2016, 49, 340–345 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structure, HRTEM, STEM, STEM-EDXS, differential capacity plot. See DOI: 10.1039/c8ta06983b |
| This journal is © The Royal Society of Chemistry 2018 |