
Chemomechanical behaviors of layered cathode materials in alkali metal ion batteries

Abstract
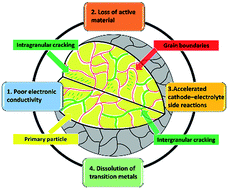
Layered cathode materials (LCMs), because of their high energy density and relatively stable performance, represent an important class of cathode materials for alkali metal ion (e.g., Li+ and Na+) batteries. Chemomechanical behaviors of LCMs, which affect battery performance dramatically, have drawn extensive attention in recent years. Most chemomechanical processes have some common chemical and structural origins that are at the center of materials chemistry, for example, defects and local bonding environments in the solid state. In this review, we first discuss the chemomechanical breakdown of LCMs by introducing their categories and negative effects on the battery performance. We then systematically analyze factors that govern the initiation and propagation of chemomechanical breakdown and summarize their formation mechanisms. Strategies that can enhance the chemomechanical properties of LCMs or reduce the destructive effects of chemomechanical breakdown are then discussed. Finally, light is shed on the new state-of-the-art techniques that have been applied to study chemomechanical breakdown. This review virtually includes most aspects of the chemomechanical behaviors of LCMs and provides some insights into the important chemical motifs that determine the chemomechanical properties. Therefore, we believe that advanced design protocols of LCMs can be developed to effectively address the chemomechanical breakdown issue of LCMs.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

