
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of magnesiothermic reduction of silica to porous silicon for lithium-ion battery applications and beyond†
Jake
Entwistle
,
Anthony
Rennie‡
 and
Siddharth
Patwardhan
*
and
Siddharth
Patwardhan
*
Department of Chemical and Biological Engineering, The University of Sheffield, Mappin Street, Sheffield S1 3JD, UK. E-mail: s.patwardhan@sheffield.ac.uk
First published on 11th September 2018
Abstract
Increasing demands for portable power applications are pushing conventional battery chemistries to their theoretical limits. Silicon has potential as an anode material to increase lithium-ion cell capacity. The associated volume change during lithiation/delithiation leads to a decline in capacity during cycling and low lithium diffusion rates within silicon limit high rate performance. Porous silicon can potentially address the poor cyclability and rate capabilities simultaneously by minimising stresses and providing smaller silicon substructures for lithium diffusion. Template assisted synthesis and magnesiothermic reduction of silica to silicon offers a facile and scalable route for the production of porous silicon structures even when using a non-porous feedstock. This review collates the available literature concerning the effects of reaction conditions through the reduction reaction. We highlight that it is important to report in detail all reaction conditions and complete characterisation of both the reactant and the product. The battery performance of these porous silicon structures is discussed and future research directions are identified. These outcomes will enable the identification of a clear design pathway for the bespoke production of porous silicon.
 Jake Entwistle, Anthony Rennie and Siddharth Patwardhan | Jake Entwistle (left) is currently working towards his PhD at the University of Sheffield at the Energy Storage and Applications centre for doctoral training. He completed MChem in 2015 at the University of Sheffield. Jake's research revolves around energy storage, particularly the sustainable synthesis, characterisation and prototyping of next generation battery materials. Dr Anthony Rennie (centre) is a Research Scientist at Faradion Ltd. Prior to that he was a Research Fellow at the University of Sheffield, responsible for the Energy Storage lab and the development of new projects such as ionic liquid electrolytes for energy storage. He received MEng in Chemical Engineering from the University of Strathclyde and a PhD from the same department in 2011 for research on porous carbon materials for supercapacitors. Professor Patwardhan specialises in the materials chemistry–chemical engineering interface, with a focus on sustainable and scalable materials and processes for application in energy, environmental, biomedical and engineering sectors. He received a Chemical Engineering degree from the University of Pune and PhD (2003) in materials science from the University of Cincinnati, followed by post-doctoral experience in inorganic chemistry at Nottingham Trent University. After working as a Lecturer at the University of Strathclyde in Chemical Engineering, he moved to Sheffield as a Senior Lecturer and subsequently was promoted to a personal chair in 2018. |
1 Silicon anodes
Rechargeable batteries play an important role in portable electronics and are increasingly being incorporated on a larger scale into electric transportation. Lithium-ion batteries (LIB) have become the chemistry of choice due to their combination of good energy and power density.1,2 The development of lithium-based rechargeable batteries with high energy and power density, improved safety and low cost is highly desirable.3 Silicon offers a significant volumetric and gravimetric energy density advantage over conventional LIB anode materials such as graphite. At room temperature the theoretical specific capacity of Si is 3580 mA h g−1 (and 2190 mA h ml−1)4 corresponding to the formation of the Li15Si4 phase. This is substantially larger than that of graphite 371 mA h g−1 (830 mA h ml−1). Silicon's low operational voltage, high earth abundance and low toxicity are additional benefits.In conventional electrode materials, lithiation and delithiation follow an intercalation mechanism where lithium ions are inserted and extracted from interstitial sites of the host material. This results in relatively small structural changes and good capacity retention. Silicon, in contrast forms an alloy with lithium, which involves the breaking and reforming of chemical bonds with the host structure upon every cycle. The large number of lithium ions inserted into silicon causes large volume changes up to 280%.5 Such a volume change within composite electrodes leads to structural damage, isolation of active material and ultimately loss of capacity. This poses a significant challenge for the development of long cycle life high capacity silicon anodes.
1.1 Understanding silicon lithiation
At room temperature, lithiation does not occur spontaneously according to the thermodynamic phase diagram.6 Silicon can either be crystalline or amorphous prior to lithiation. If silicon is crystalline then the first lithiation is via a two-phase mechanism between Si and amorphous LixSi phases which are separated by a short reaction front of a few nm.7 There is a large activation energy required to break Si–Si bonds within the crystalline silicon matrix and therefore a high concentration of lithium ions is required to weaken the Si–Si bonds at the reaction front, leading to favourable lithiation kinetics and two-phased behaviour.7,8 This amorphous LixSi phase is found to crystallise to Li15Si4 around 100 mV vs. Li/Li+ (ref. 7); however, this may be higher depending on factors such as the crystallite size.9 This results in a galvanostatic voltage profile with a voltage plateau around 0.1 V vs. Li/Li+, suggesting that a two-phase mechanism dominates the lithiation process.During the first delithiation another two phase mechanism occurs in reverse, eventually yielding amorphous silicon. After the first cycle, lithiation subsequently occurs into amorphous Si. The conversion of crystalline silicon through lithiation over the initial cycles was studied through in situ XRD6 and a physical model of amorphous silicon lithiation has been developed based on 7Li NMR investigations.8In situ TEM observations for lithiation of both crystalline and amorphous silicon indicate a critical size for nanoparticles, beyond which they crack, 150 nm diameter for crystalline silicon and 870 nm for the amorphous phase.10,11 Volume changes of up to 300% in magnitude have been studied in silicon thin films with AFM and were shown to be roughly linear with Li content,12 with the linear increase with the lithiation level also being modelled.4
Within the constrained environment of a composite electrode, the volume expansion of silicon becomes an issue for three main reasons as depicted in Fig. 1.13 Firstly considering Fig. 1(a), stresses within individual particles lead to fracturing and eventually pulverisation. Fig. 1(b) shows that the impingement of expanding material in the electrode can lead to fragmentation on a larger scale, disconnecting sections of the electrode. In each case, active material becomes electrically isolated and no longer contributes to the electrode capacity. Fig. 1(c) illustrates the case when constant expansion and contraction leads to the cracking of the Solid Electrolyte Interphase (SEI) layer, exposing fresh active material and causing further breakdown of the electrolyte. The thickening of the SEI can increase the internal resistance of the cell and consumes lithium from the electrolyte.
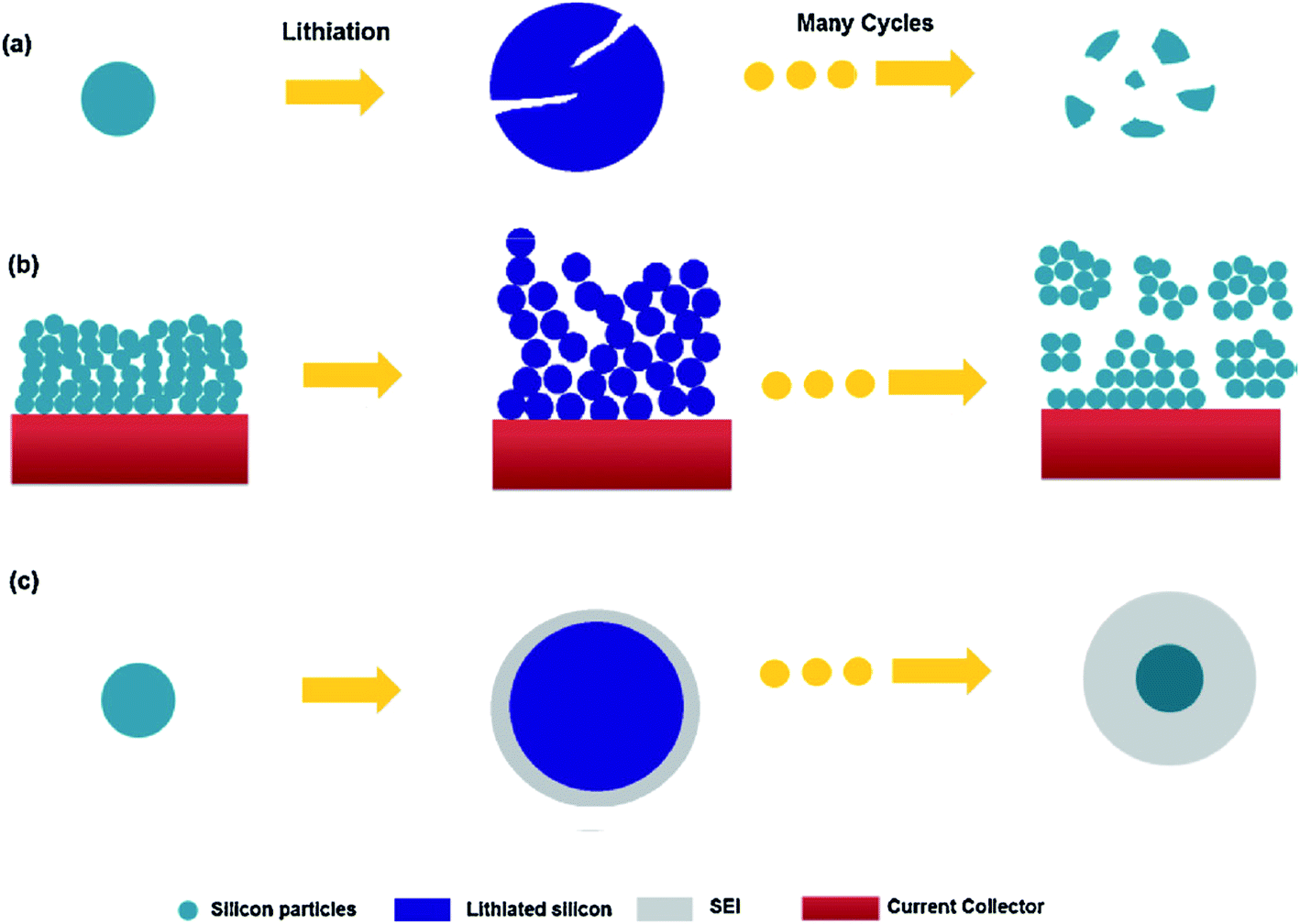 | ||
| Fig. 1 Electrode failure mechanisms for silicon active material: (a) material cracking and pulverization. (b) Electrode expansion and impingement leading to isolation. (c) Continual SEI formation.13 Image reproduced with permission. | ||
Another challenge is the relatively low lithium diffusion rates within silicon, 10−10 to 10−11 cm2 s−1,14,15 compared with the range 10−6 to 10−11 cm2 s−1 reported for graphite electrodes.16–18
During lithiation, the process is complete when the lithium-rich Li15Si4 phase is formed at the surface of the silicon electrode; similarly delithiation finishes when the lithium ions are extracted from the very outer surface layer. This arises as the potential of the cell is determined by the chemical potential difference of lithium ions between the two electrodes. If the potential drops below the operational voltage window, then regardless of whether inner silicon has participated in lithiation/delithiation, the process will be stopped. This low diffusion rate poses a kinetic barrier to achieving the theoretical capacity of silicon and is pronounced with larger silicon grain sizes. At higher current rates, the voltage drop across the internal resistance of the battery can compound, leading to the theoretical capacity not being reached.19
1.2 The role of porous silicon
Porous morphologies can potentially address the challenges of volumetric expansion and slow lithium diffusion. Porous silicon can expand into its own pore volume, thus limiting stresses on the material. Stress generation upon lithiation has been modelled for lithium insertion materials20,21 which in turn have been applied to models of lithiation in porous silicon structures.19,22,23 A comparison of hollow and solid amorphous silicon nanospheres of the same silicon volume modelled by Yao et al. showed that the maximum tensile stresses experienced by a hollow sphere is significantly lower vs. lithiation time, 83.5 vs. 449.7 MPa respectively (Fig. 2(A)).22 Ge et al. simulated the effect of pore size and porosity in the range of initial pore sizes 1–9 nm, showing that lower porosities lead to higher induced stress and smaller pore sizes result in higher maximum hoop stresses around the pore (Fig. 2(B)).19 Li et al. also showed a linear decrease in the porous particle volume change by increasing the void volume fraction for materials containing 10 and 25 nm radius ordered pores. Fig. 2(C) shows that increasing the void fraction to 60% should keep the overall particle expansion within the narrower volumetric expansion range of 75 to 150%.23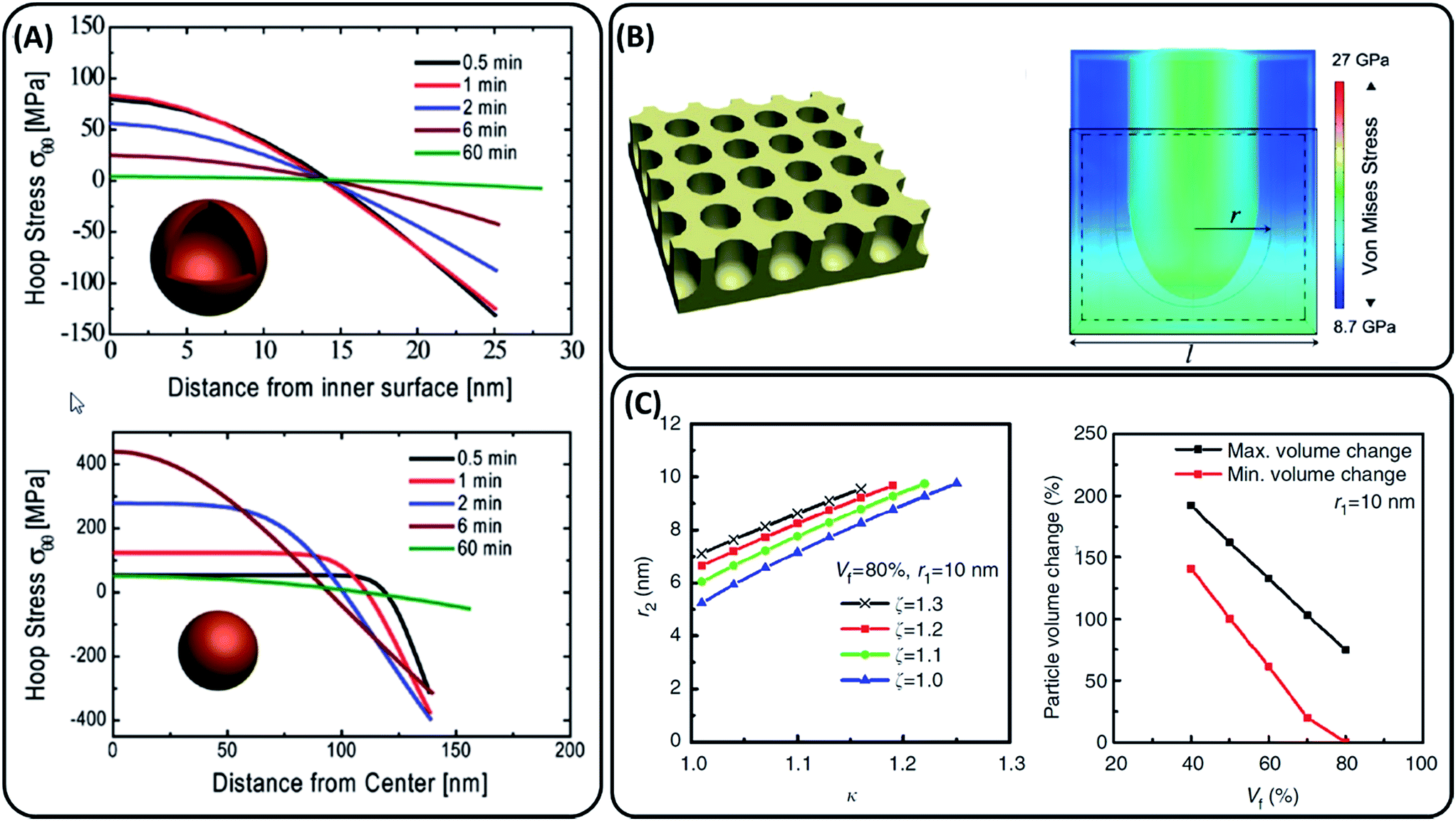 | ||
| Fig. 2 (A) Stress evolution during lithiation in a hollow sphere vs. a solid sphere with the same volume of silicon reported by Yao et al. Positive stress values are tensile stress, while negative are compressive.22 (B) Schematic of the porous structure modelled by Ge et al. with cross-sectional analysis of stress generation under lithiation.19 (C) Expansion rates of a mesoporous particle along the radial direction (κ), axial direction (ζ) and pore radius after lithiation (r2) reported by Li et al. Particle volume expansion versus void fraction (Vf).23 Each image has been reproduced with permission from respective references. | ||
Porous silicon with a high surface area can increase the accessibility of the electrolyte to silicon surfaces, shortening lithium diffusion lengths and increasing available capacity at higher rates. Porous structures with thin walls and silicon substructures can shorten the diffusion length of lithium within silicon. Polycrystalline porous structures with small silicon domains are hypothesised to have a higher resistance to fracture during lithiation similar to the observed strong size dependence of fracture of silicon nanoparticles.10 An in situ TEM observation has shown that the critical particle diameter for fracture in porous particles reaches 1520 nm.9 Additionally, in situ TEM and dynamic simulation of the lithiation behaviour of porous silicon particles found that the smaller domains of porous particles disfavour the crystalline c-Li15Si4 phase upon full lithiation, providing a more favourable stress evolution on expansion (Fig. 3).9 Additionally, it was shown that these porous particles lithiated in an end-to-end fashion (Fig. 3(e–h)), as opposed to larger nanoparticles which lithiated in a surface-to-centre manner (Fig. 3(a–d)).9
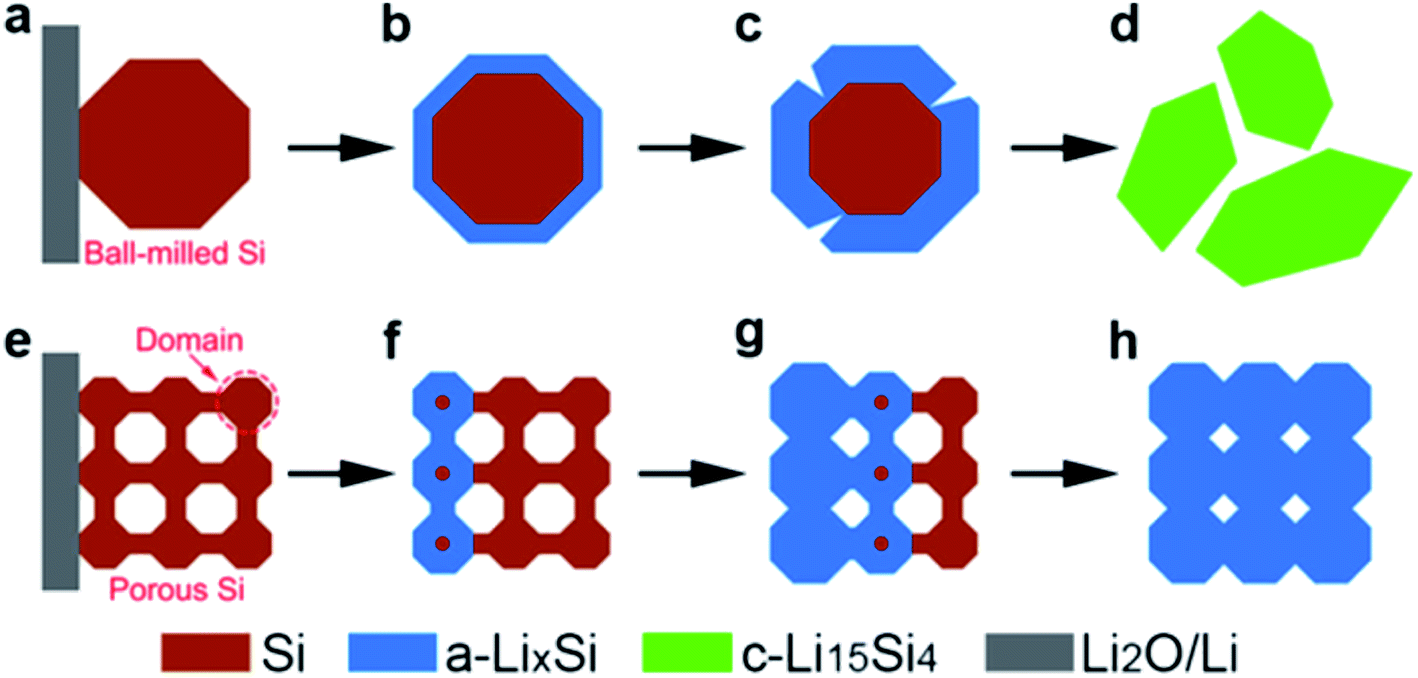 | ||
| Fig. 3 Schematic for the lithiation of ball-milled silicon and porous silicon nanoparticles. (a–d) Surface-to-center lithiation of ball milled silicon. (e–h) End–end lithiation manner of a porous silicon particle.9 Figure reproduced with permission. | ||
High surface area porous silicon structures will however generate a larger SEI simply because of the increased electrode area in contact with the electrolyte. This could be a significant drawback when coupled with the volume expansion of silicon and relative instability of the SEI. Surface coatings and particle encapsulation mitigate these issues and are described in Section 3.
1.3 Synthesis/fabrication of silicon
The worldwide annual production of silicon metal amounted to 8100 tons in 2015.28 The industrial scale synthesis of metallurgical grade silicon is typically achieved by the reduction of silica with carbon in electric arc furnaces at temperatures over 2000 °C.29,30 A common industrial method of refining metallurgical grade silicon is chemical vapour decomposition (CVD), see Fig. 4(b). CVD methods usually require reaction temperatures of 1100 °C in the presence of hydrogen gas. Producing the volatile silicon precursors needed also requires a reaction with hydrochloric acid at 350 °C.29 The other major refinement methods rely on the crystallisation of molten silicon, again requiring high temperatures >1414 °C to achieve molten silicon, Fig. 4(a). The development of environmentally friendly, low cost and scalable production processes for high performance silicon anodes is essential.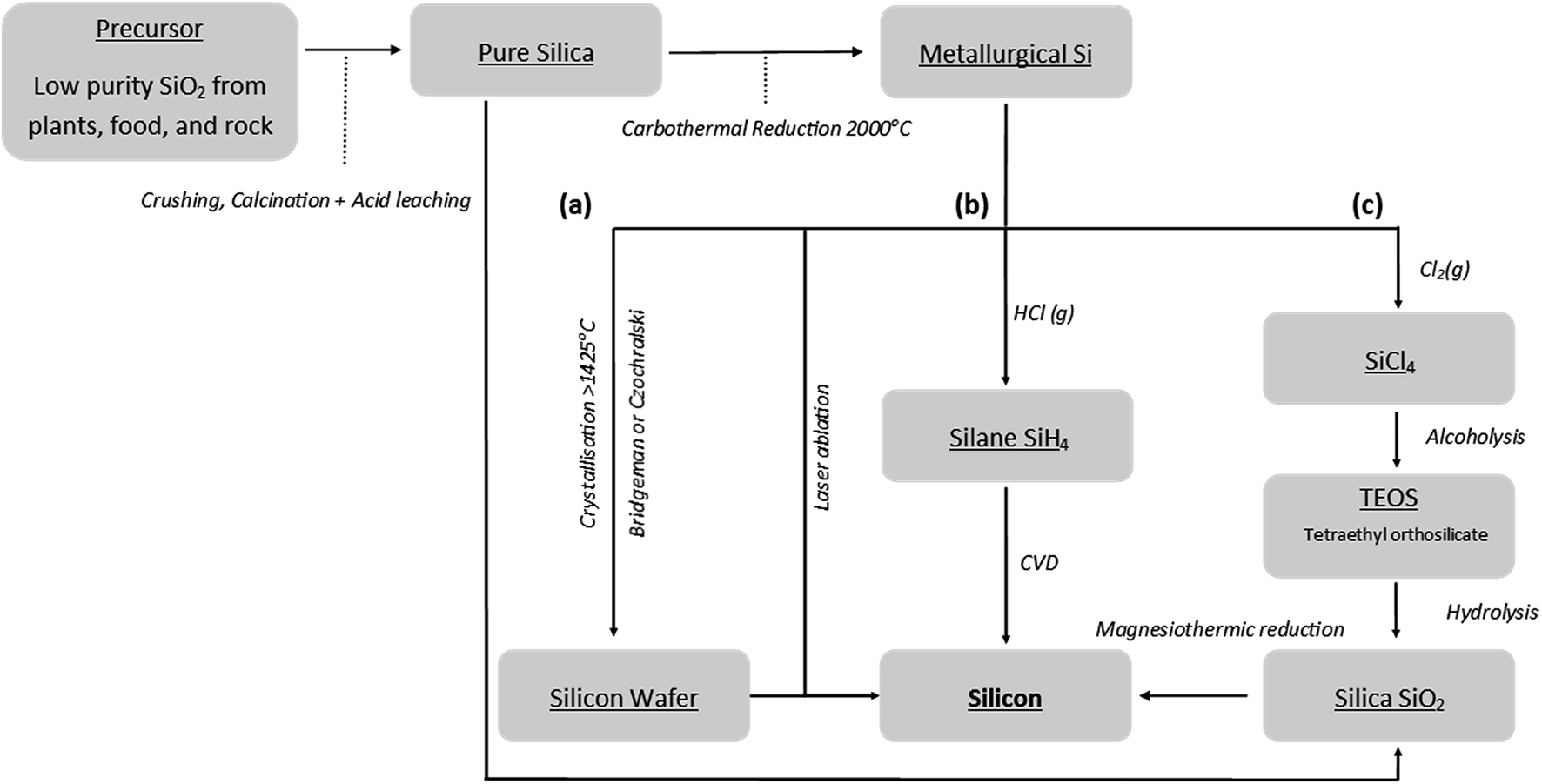 | ||
| Fig. 4 Flow chart of existing bulk silicon synthesis routes including magnesium reduction. | ||
There has been great progress in developing silicon anodes with a wide range of nano-structuring techniques being employed and extensively reviewed elsewhere.13,24–26 High capacities and good cyclability have been demonstrated using nanoscale silicon anode structuring.13,24,26 Porous silicon structures have also been reviewed by Ge et al.25 However, nanoscale engineering often requires a high degree of precision synthesis and can involve aggressive reaction conditions. Using energy intensive synthesis techniques will likely lead to high cost of materials and the inability to produce materials on an industrial scale.27
Magnesiothermic reduction can offer a facile method for the bulk synthesis of porous silicon with varying degrees of structural control. Fig. 4 highlights the benefits of this single step reduction method from silica to silicon in comparison to existing technologies. This review collates the available literature concerning the influence of reaction conditions on the magnesiothermic reduction reaction as well as highlighting gaps in the current understanding. The use of porous silicon produced through magnesiothermic reduction as an active material for anodes in LIBs is the focus of this review, but potential applications may be much further ranging with impact in fields such as photoluminescence,31 solar power,32 photocatalysis,33 drug delivery34 and catalysis support.35
2 Magnesiothermic reduction
2.1 Background
Magnesiothermic reduction of silica (SiO2) has the potential to produce porous silicon materials at lower temperatures than conventional silica reduction methods.36 As the melting point of silicon is 1414 °C, carbothermal reduction at 2000 °C is not suitable for maintaining the silica template structure to give silicon pseudo-morph analogues. Magnesiothermic reduction has been demonstrated to produce silicon structures from silica in the temperature range of 500–950 °C, permitting template assisted design of silicon structures. This method has shown the ability to preserve intricate features in the silicon produced as small as 15 nm.37 The high diversity and robust understanding of silica chemistry allow for the possibility of creating a wide range of silica template structures with tailored geometries. This reduction entails the reaction of magnesium with silica resulting in an interwoven composite product of magnesia (MgO) and silicon (reaction (1)).| SiO2 + 2Mg → Si + 2MgO | (1) |
Magnesia is easily removed with HCl, leaving a silicon replica behind that possesses a higher surface area than the starting template. Fig. 5 summarises the two step reduction–etching process. The interwoven nature of this morphology is crucial to allow the removal of magnesia in this way. The formation of the interwoven aggregate morphology of the product Si and MgO is thought to relate to the stability of the reaction interface and flux of reactants across the product phases.38
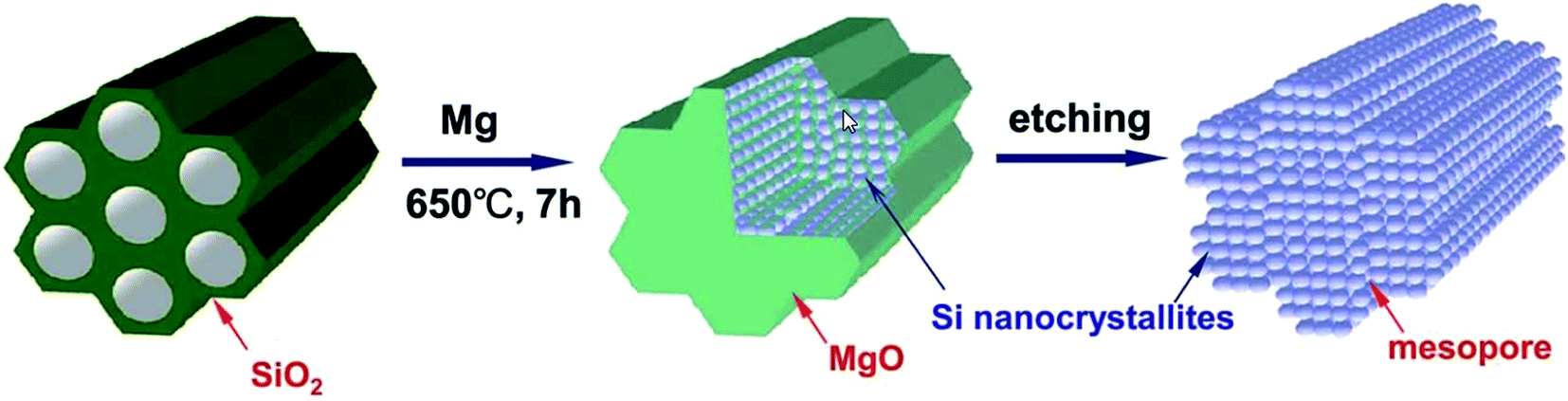 | ||
| Fig. 5 Illustration of the magnesiothermic reduction process to produce porous silicon from silica.39 Figure reproduced with permission. | ||
The Gibbs energy of magnesiothermic reduction is negative for the entire temperature range 0–1000 °C and this indicates that the reaction is exergonic.40,41 The enthalpy of reaction (1) is exothermic and has significant ramifications as discussed below.41 The melting point of magnesium is 650 °C, and the vapour pressure of Mg at 428 °C is 1 Pa. This enables solid magnesium and silica to be placed separately in the reaction vessel and the magnesium gas to diffuse to reduce the silica and/or liquid magnesium to flow over the silica.36,42
It is important to note the side reaction can reduce the yield of silicon through the formation of magnesium silicide (reaction (2)). A number of studies when placing the magnesium and silica separately and relying on the gas–solid type reaction report the silica close to the source being reduced to Mg2Si and a middle region forming Si, with further displaced sample showing no reduction and remaining as SiO2.36,40,42,43 Mg2Si formation reduces the yield of the reaction and affects the product morphology upon removal which occurs simultaneously with MgO removal in the HCl wash. Mg2Si is not thermodynamically stable in the presence of SiO2 and therefore its formation is due to kinetic limitations.44 Silanes are produced by the reaction of magnesium silicide with acid. They react spontaneously with air.
| Si(s) + 2Mg(g) → Mg2Si(s) | (2) |
Fig. 6 displays key reaction variables with respect to the stages of the reaction. Below we aim to summarise and evaluate critically the literature findings (ESI Table S1† displays in detail the key parameters identified in the literature).
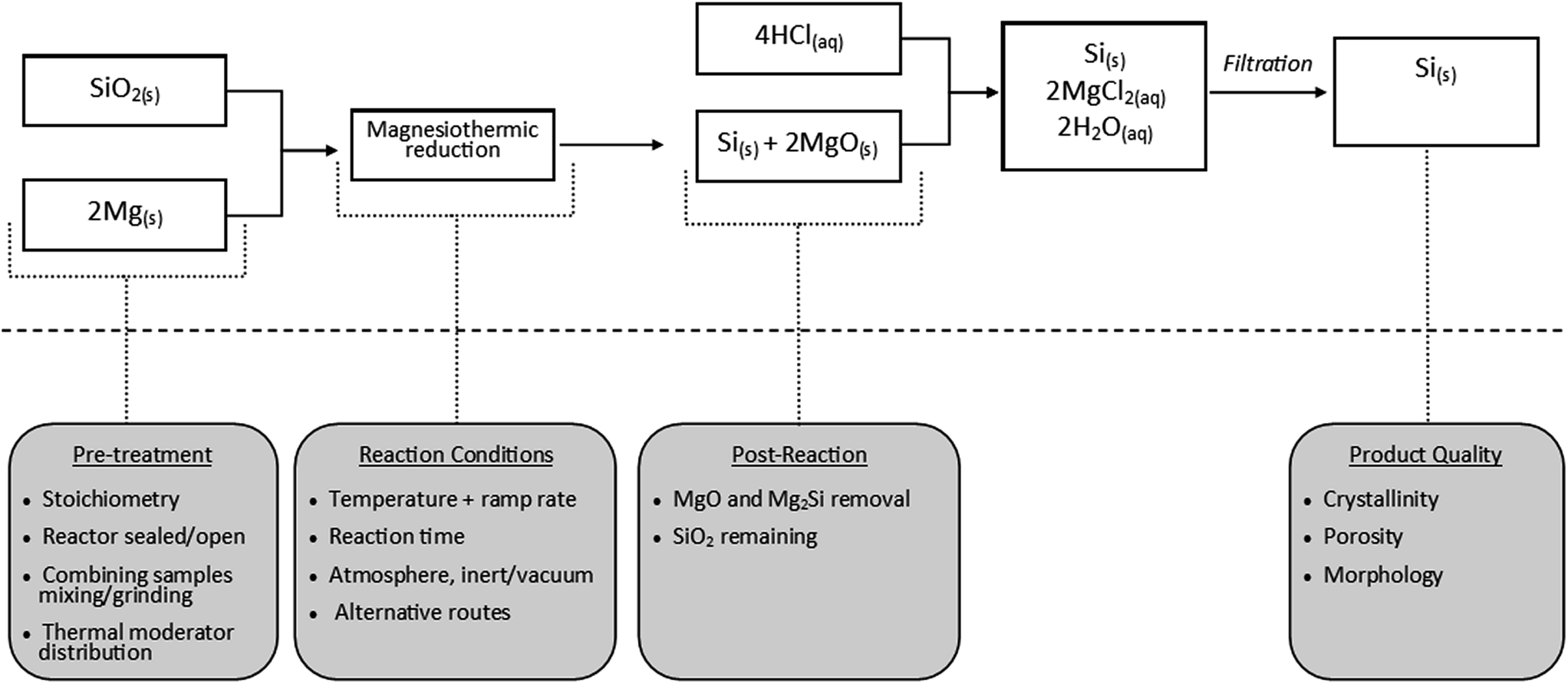 | ||
| Fig. 6 Scheme outlaying the magnesiothermic reduction and summary of key design factors for consideration. | ||
A number of challenges remain in the development of a controllable magnesiothermic reduction method for porous silicon production, primarily the preservation of the silica template morphology in the silicon analogue, which underpins the rationale of the method. Another challenge is control over the extent of the reaction to fully reduce the silica avoiding the need for the undesirable process of HF etching, while simultaneously maintaining the silicon yield by avoiding overreduction to form Mg2Si.
In order to design the porous silicon product that is desirable for anode applications, the feedstock and the process parameters governing the magnesiothermic reduction process and their effects on the properties and performance of silicon should be understood. In Section 2.2 we discuss these key features and their effects on the product silicon.
As evident from the foregoing, a truly comparative literature review is difficult as most studies reduce just one specific silica material under set reaction conditions and report a limited number of variables. Magnesiothermic reduction reaction conditions are not all comparable from one study to another. As a result, the silicon product morphology can vary for the same silica templates between studies.
2.2 Reduction conditions
The key control parameters for the reduction reaction are: reaction time, temperature and temperature ramp rate, reactant proximity and morphology, and molar ratio of reactants. Conventionally, magnesiothermic reduction is carried out under a flowing 95% argon or a nitrogen atmosphere, although some studies have performed the reduction under vacuum to promote a higher vapour pressure of magnesium.45–47 Another source of magnesium gas can also be achieved from the volatilization of Mg2Si.48,49 All these parameters have significant effects on the silicon product. However, a comprehensive study on the effect of reaction conditions on the product properties is lacking in the literature. In Section 2.2 below we aim to highlight key studies where quantitative conclusions can be drawn between reaction conditions and product silicon properties.Liu et al.42 studied the effect of the temperature ramp rate up to 650 °C in a closed type reactor of Mg powder and rice husk derived silica (Specific Surface Area, SSA 289 m2 g−1, and Specific Pore Volume, SPV 0.45 cm3 g−1). They compared the difference between 5 °C min−1 and 40 °C min−1 heating rates on the morphology of the silicon produced from magnesiothermic reduction. The higher ramp rate of 40 °C min−1 produced mainly macroporous silicon particles with a SSA of 54 m2 g−1 and negligible pores below 20 nm, while the material produced at 5 °C min−1 gave partially interconnected mesoporous silicon nanoparticles with a SSA of 245 m2 g−1. The 5 °C min−1 sample had significant porosity around 10 nm width similar to the template, and pores in the 20–30 nm range which contributed to the increase in the pore volume to 0.74 cm3 g−1. Liu et al. validated this study with Thermal Gravimetric Analysis (TGA) and Differential Thermal Analysis (DTA) showing a strongly exothermic reaction occurring at 400–450 °C. As no significant reaction occurs below 400 °C, quick ramping can be used to reach this point. Further decreasing heating rate below 5 °C min−1 does not decrease the particle size or crystallinity of the obtained particles. Upon scaling the reaction volume up to 5 g, a 1 °C min−1 heating rate was used.
Consistently the study of Shi et al.50 investigated the effect of the heating rate on silica derived from rice husks at 5, 3 and 1 °C min−1 for a 4 g reaction volume where a substantial effect on the morphology was observed. The higher rates were observed to fuse silicon particles together and remove micropores, in turn decreasing the pore volume and surface area. Only the sample product at 1 °C min−1 was slow enough to prevent significant changes in the morphology. An XRD study on the heating rate also confirmed the formation of larger crystallite sizes for MgO and Si at faster ramping rates.42,50 A higher ramping rate also increases the presence of Mg2Si as discussed in Section 2.2.2.
Shi et al.50 also found that faster ramping rates and the associated heat accumulation lead to increased Mg2SiO4 formation, and in this case the runaway kinetics of the faster ramping rates could have caused local mismatches at the SiO2/Mg interface leading to more Mg2SiO4 formation, as further discussed in Section 2.2.4.
Barati et al.32,47 report that between 750 °C and 950 °C, temperature has little effect on the Si yield. However, they reported that when the Mg stoichiometric ratio is increased above 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, higher temperatures yield more silicon. It is shown that at 750 °C significantly more Mg2Si was produced than at 950 °C (discussed below Section 2.2.4). This effect is attributed to higher temperatures accelerating the kinetics of the solid state reduction of SiO2 by Mg2Si, as described in reaction (3). This solid state reduction is diffusion controlled so longer times and higher temperatures favour reaction (3) and increase the silicon yield.32
:1, higher temperatures yield more silicon. It is shown that at 750 °C significantly more Mg2Si was produced than at 950 °C (discussed below Section 2.2.4). This effect is attributed to higher temperatures accelerating the kinetics of the solid state reduction of SiO2 by Mg2Si, as described in reaction (3). This solid state reduction is diffusion controlled so longer times and higher temperatures favour reaction (3) and increase the silicon yield.32
| SiO2 + Mg2Si → 2Si + 2MgO | (3) |
In addition, Xie et al.51 showed that Mg2SiO4 formation can be reduced by increasing the reaction temperature from 700 °C to 900 °C. Increasing the reaction temperature increases Mg vapour pressure and hence concentration at the SiO2/Mg interface favouring MgO formation over Mg2SiO4.
A recent study has shown that the surface area and pore volume decrease as the reaction temperature is increased between 500 and 800 °C and an increase in the average mesopore diameter is observed. This effect is determined to be from a higher degree of structural damage of the template at higher temperatures.52
A study of reaction temperature and time by Liu et al.40 was conducted on nonporous silica with a bundled nanowire morpology. They report intuitively that higher temperatures lead to silicon at lower reaction times. Interestingly, by initiating the reaction at 600 °C and then reducing the temperature to 400 °C, crystalline silicon was formed, whereas at 400 °C reaction temperature no silicon was observed even with ‘overnight’ reaction times. The onset of the reduction reaction has been reported between 400 and 540 °C (depending on the silica source),32,42 explaining the need for this initiation temperature.
The study by Wu et al.46 found that reaction times of 2 and 5 hours had little effect on the morphology of the product silicon and more so the state of magnesium, gaseous or liquid, during the reaction. The promotion of the gaseous state of Mg is achieved by applying a vacuum to the reaction.46 This could however further indicate the reaction reaching an equilibrium state in under 2 hours.
:Si above the stoichiometric level of 2:1 decreases the silicon yield by increasing the formation of Mg2Si. XRD quantitatively showed that an excess of magnesium of 5 wt% (2.1:1 Mg:SiO2 stoichiometry) gave the maximum yield of silicon.32,47
Magnesium silicate Mg2SiO4 has been detected in a number of XRD studies. Mg2SiO4 is not easily removed with HCl, and therefore it will affect the silicon product properties. Insufficient Mg at the SiO2/Mg reaction interface also favours Mg2SiO4 formation.32 This is supported by Chen et al.54 where increasing the Mg molar ratio reduced the formation of Mg2SiO4 even at relatively low temperatures.
The mixing of the reactants by a ball milling process can provide enough kinetic energy to reach the reaction ignition point, and has been used to perform the magnesium reduction. This method has also been scaled up to a 5 litre scale in an attrition mill.55 Care should therefore be taken when using milling to mix reactants.
The heat released during the reaction requires lower ramping rates for larger batch sizes as there is more heat accumulation in these reactors.42 Batchelor et al.56 demonstrated the production of mesoporous silicon from biosilica sources on a 30 g reaction scale. The key to this larger batch size was the use of a thermal moderator, sodium chloride (NaCl), which prevents silicon particles from sintering together. Luo et al.43 also reported the use of NaCl as a heat scavenger in the reduction reaction which showed the ability to better maintain the diatom biosilica structure in the reduced form. The crystallite size was also reduced as a result of reducing the reaction temperature.43
As the reaction temperature begins to rise above the set temperature, NaCl melts at 801 °C, consuming excess heat due to its high enthalpy of formation. Luo et al.43 ground Mg and SiO2 together, reacting in a sealed vessel for a batch size of 1.9 g, typical of many reported synthesis routes. Without thermal mediators they reported true reaction temperatures above 1300 °C, and with their mediators the temperature was kept between 840 and 1100 °C. Greatly increasing the ratio of NaCl was unable to keep the reaction temperature below 801 °C as the method is limited by the degree of mixing between NaCl and silica. Others have shown more elaborate NaCl surface coating of silica to be beneficial.57,58 The addition of NaCl has also been reported to reduce the presence of Mg2Si in the product silicon.59
Interestingly Xing et al.62 report smaller crystallite sizes (19.9 nm) when the silica template had a higher porosity and higher surface area. This is attributed to the hindrance of mass transport during heat treatment retarding the coarsening of the silicon crystallites. Silica feature size affects the crystallite formation with smaller features leading to smaller silicon crystallites. A lower limit of ∼13 nm has been suggested for the crystallite size relating to the critical nucleation size for silicon under typical reduction conditions (675 °C 2 h).37 However, for smaller template features the smallest crystallite sizes of 4 nm have been reported.60
Magnesia removal is attributed to the increase in pore volume upon HCl etching. Conclusions have been drawn from the MgO crystallite size before etching and the introduction of pore volume in an attempt to correlate MgO crystallite size and pore diameter. However a direct correlation has not been found.61,62
| 4Mg(g) + SiO2 → 2MgO + Mg2Si | (4) |
| Mg2Si + O2 → 2MgO + Si | (5) |
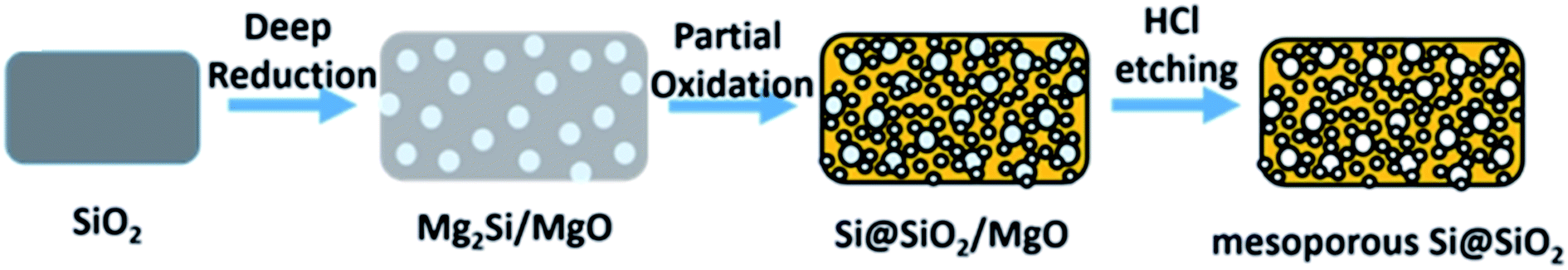 | ||
| Fig. 7 Schematic illustration of the deep reduction and partial oxidation process. Reprinted with permission from ref. 63. Copyright (2016) American Chemical Society. | ||
They further optimised the oxidation time and temperature in ref. 64. Using commercial silica they produced a ‘nanoporous’ high surface area product with a significant batch size (10 g of product) with a yield above 90%. An excess of Mg to Si at 500 °C for 10 hours propagated the Mg2Si intermediate formation which in a second step was oxidised in air. Bulk silicon oxidation was not significant below 800 °C, but a surface SiO2 layer was produced. This reduction method, with a high yield and control on converting Mg2Si, could have great potential for silica reduction. The formation of surface oxide affords improved cycle life by potential of improving the quality of the SEI layer formed. This is consistent with similar literature studies.65,66
A two step synthesis used by Choi et al.67 was demonstrated where first aluminothermic reduction (reaction (6)) at 900 °C and then subsequent magnesiothermic reduction at 700 °C of diatom silica produced porous silicon (reaction (7)). The two steps involved in this synthesis each have lower Gibbs energies which avoided heat accumulation during reduction.41 By controlling the stoichiometry in the magnesiothermic reduction, varying amounts of Al2O3 remained in the product. Retaining Al2O3 in the final product reportedly improved the cycle life of the silicon materials aiding stable SEI formation and buffering volume expansion similar to that reported by Liang, et al.63 Since Al2O3 does not contribute to the capacity of the electrode, the specific capacity of the material was reduced.
| 3SiO2 + 4Al → 2Al2O3 + 3Si | (6) |
| Al2O3 + 3Mg → 3MgO + 2Al | (7) |
Expanding the field of metallothermic reduction, Lai et al.52 combined aluminothermic and magnesiothermic reduction into a one-step synthesis route. A eutectic mixture of 70% magnesium and 30% aluminium was used allowing the reduction to proceed at 450 °C. The silicon formed by this method was amorphous, which could be significantly beneficial for lithium-ion battery applications.11
2.3 Precursor silica
A wide range of biological and synthetic silica sources are available and have been reduced via magnesiothermic reduction for LIBs. Both sources, as shown in ESI Table S1,† have the ability to produce porous silicon with a variety of pore properties. It should be noted that biologically derived silica such as rice husk and bamboo silk requires the removal of organic components and commonly needs acid leaching of metal impurities. Synthetic silica sources can be equally time consuming as well as resource and energy intensive to produce (see Fig. 4(c)). In the striving to produce battery materials in a more economical and environmentally friendly way, the choice of silica template is also significant.Table S1† summaries the key parameters of silica before and silicon after reduction where these data were available. In general, many of the reduction reactions produce mesoporous silicon with a range of surface areas between 24–350 m2 g−1 and pore volumes 0.11–1.1 cm3 g−1. Interestingly these mesoporous properties also appear in samples which initially were non-porous such as sand59 and silica spheres.51,52 In these cases the nature of the silicon-magnesia interwoven aggregate introduces porosity to the structure through the removal of the magnesia phase. The same effect is seen on non-porous starting silica/silicon used in the ‘deep reduction partial oxidation’ method.63,64 It appears that when the overall template morphology is maintained, and if reaction temperatures can be controlled sufficiently below the silicon melting point, pores in the mesopore region and overall pore volumes will increase irrespective of the porosity of the precursors. This is somewhat expected as the magnesia phase occupies ∼65% of the volume in the product structure and oxygen is being removed from the initial template.48
Rice husks have been reduced by magnesiothermic reduction in a number of studies.42,50,61,62,68 Table S1† shows how the initial rice husk precursors have similar SSA and pore volumes, but the purity of the silica varies. Where reported, upon reduction, the pore volumes increase in all studies with surface areas remaining similar to or lower than that of the starting silica, with the exception of ref. 61 where the surface area and pore volume are dramatically decreased from 234 m2 g−1 to 42 m2 g−1 and 0.43 cm3 g−1 to 0.31 cm3 g−1. In that study, a lower reaction temperature of 500 °C may contribute to the pore properties not following the trend. Note that this is below the onset temperature reported at 540 °C by Larbi et al.32 who also reduced silica rice husk. This example highlights the lack of understanding in the evolution of magnesiothermic reduction reactions. Additionally, this shows how specific factors in each case can also contribute to significant variation in product properties; these factors include furnace design, crucible design and packing and mixing of reactants.
Table S1† shows a number of examples where the magnesiothermic reduction conditions have had a much greater effect on the silicon morphology than the template used.46,69,70 Kim et al.71 utilised a change of morphology to obtain their desired structure; they reduced vertically aligned mesoporous silica to obtain 10 nm silicon nanoparticles dispersed on graphene sheets. Similarly, Zhu and Wu utilised magnesiothermic reduction to produce silicon nanoparticles on graphene sheets.72,73 Although not initially the function of this method, it has proven to produce novel materials with good properties for lithium-ion battery applications and potentially beyond.
2.4 Summary
In this review it has been clearly shown how individual reaction parameters can affect the silicon product properties. In reality the interplay of reaction parameters with each other may add more complexity to the relationship between reaction conditions and product properties. However, the scientific understanding of the key parameters collated above should be able to provide valuable insight into the importance of reaction conditions on product properties. Table 1 below summarises the key effects of the ramp rate, temperature, time and molar ratio.| Experimental factor | Reported range | Reaction consequence |
|---|---|---|
| Ramp rate | 1–40 °C min−1 | • Magnesium reduction has severe heat accumulation, raising temperature above silicon's melting point, this has strong dependence on ramp rate and batch size |
| • Severe heat accumulation associated with faster ramp rates and larger batch sizes has shown across studies to produce a macroporous silicon product. This product does not maintain the silica template structure and typically has a lower surface area and pore volume compared to silicon produced at a lower ramp rate | ||
| • Slower ramping rates mitigate heat accumulation and are increasingly necessary for larger batches | ||
| • Heat Scavengers such as NaCl have shown to be beneficial for limiting heat accumulation allowing faster ramp rates | ||
| Reaction temperature | 500–900 °C | • Heat accumulation may lead to true reaction temperatures higher than set conditions |
| • The onset of reduction occurs between 400 and 540 °C | ||
| • Higher temps have been shown to decrease Mg2Si and Mg2SiO4 formation increasing the silicon yield | ||
| • Higher temperatures and longer reaction times are the drivers for increasing the silicon crystallite size | ||
| Reduction time | 0.5–12 hours | • Silicon yield has been shown to plateau beyond reaction times of 2 hours |
| • Reduction time has scope for further optimisation as it has been indicated that the reaction can be initiated at higher temperatures which is then greatly reduced until completion | ||
| Molar ratio | 1.5–3 | • If a ratio less than the stoichiometric ratio is used, 2:1, Mg:SiO2 silica remains in the product |
| • Slightly excess Mg gives a higher silicon yield | ||
| • Further increasing the magnesium molar ration decreases Mg2SiO4 formation but increases Mg2Si formation |
A wide variety of silica precursors have been studied through magnesiothermic reduction. By reviewing the relevant literature, it is clear that under circumstances where the heat accumulation has been mitigated, the reduction of silica will introduce mesopores into the silicon product and increase the overall pore volume. This is observed for precursors which initially have micropores and mesopores and even for non-porous precursors. A strong indication of heat accumulation, causing the reaction to approach or exceed the melting point of silicon, is a typical macroporous product with spherical pores around 200 nm.46,66 These two effects can be described by the nature of the interwoven aggregate silicon/magnesia product phase. At lower temperatures, smaller magnesia phases (in the mesopore size range 2–50 nm) are formed interwoven with silicon, with elevated temperatures causing the aggregation of magnesia crystallites into larger grains (in the macropore range ∼ 200 nm).
3 Anode performances
The studies using rice husks as a silica source presented in Table 2 are examples of how varying magnesiothermic reduction conditions (ESI Table S1†) can greatly affect the SSA and pore volumes of the silicon product. In general, lower surface areas and pore volumes result in lower capacities and poorer capacity retention.42,61,62 The example of rice husk reduction from Liu et al.42 shows the benefits of porous silicon, with a high stable capacity of 1750 mA h g−1 and reasonable capacity retention over 300 cycles. Factors such as the electrode composite and electrolyte additives also likely played a key role in performance parameters. Interestingly the best performing rice husk derived silicon electrode was formed using a polyvinylidene fluoride (PVDF) binder; which has previously been shown to be inferior to silicon.74,75 The high porosity of these samples is likely to limit the overall particle expansion, as discussed in Section 1.2, negating the need for more flexible binders.| Silica source | Silicon properties BET surface area, pore volume | Anode performance |
|---|---|---|
| Rice husks42 | 245 m2 g−1 mesoporosity, 0.74 cm3 g−1 | 1750 mA h g−1 @ 2.1 A g−1 300 cycles, 86% capacity retention |
| Rice husks62 | 150 m2 g−1 mesoporous, 0.60 cm3 g−1 | 1600 mA h g−1 @ 1.0 A g−1 100 cycles, 76% capacity retention |
| Rice husks61 | 42 m2 g−1 mesoporous, 0.31 cm3 g−1 | 1400 mA h g−1 @ 0.08 A g−1 50 cycles, 65% capacity retention |
| SBA-15 (ref. 39) | 74 m2 g−1 mesoporous, 28 nm APD 0.56 cm3 g−1 | 2727 mA h g−1 @ 4.2 A g−1 100 cycles, 53% capacity retention |
| SBA-15 (ref. 63) | 103 m2 g−1 mesoporous majority, 4–16 nm | ≈1300 mA h g−1 @ 0.36 A g−1 100 cycles, 77% capacity retention |
| Diatom (ref. 63) | 74 m2 g−1 mesoporous, ≈30 nm average pore diameter (APD) | ≈1400 mA h g−1 @ 0.36 A g−1 100 cycles, 71% capacity retention |
| Sand (ref. 63) | 23.9 m2 g−1 mesoporous, ≈30 nm APD | ≈1782 mA h g−1 @ 0.36 A g−1 100 cycles, 82% capacity retention |
| Aerogel (ref. 63) | 239 m2 g−1 mesoporous, 4 nm pores | 1782 mA h g−1 @ 0.36 A g−1 100 cycles, 82% capacity retention |
SBA-15 is a silica with mesopores, a biphasic system of ordered hexagonal arrays of pores.76 A number of studies have used a magnesiothermic reduction method with SBA-15 to achieve ordered mesoporous silicon for anodes.39,54,63 The SBA-15 used is either produced in-house39 or purchased from different suppliers34,54,63 introducing some discrepancies between studies. The best performing SBA-15 silicon analogue is reported by Jia et al.,39 especially when considering fast charging rates as shown in Fig. 8(c). The lower surface area and larger pore diameters could perhaps be responsible for this success, as they favour less SEI formation and larger pores experience smaller stresses on expansion. This material performs well at 1500 mA h g−1 for 100 cycles with 94.4% capacity retention when using a carbon coating (Fig. 8(b)). In this case the porous silicon is coated with a 4 nm layer of amorphous carbon via chemical vapour deposition filling some pores and decreasing the surface area. With the carbon coating negating the negative effects of the increased silicon surface area causing excessive SEI formation, it can be seen that this mesoporous material has very attractive properties for LIB applications. A number of studies demonstrate the attractive properties of combining a carbon coating and porous silicon in this manner.51,77–80
 | ||
| Fig. 8 (a) Voltage profile of mesoporous silicon particles with a carbon coating (mp-Si@C). (b) Cycling performance of mesoporous silicon (mp-Si), mp-Si@C and silicon nanoparticles (nano-Si). (c) Reversible capacities of mp-Si@C cycled at different current rates. (d) Voltage profiles of mp-Si, nano-Si and graphite.39 Figure reproduced with permission. | ||
The study of Liang et al.63 using ‘deep reduction and partial oxidation’ is an example for the comparison of the effects of various porous silicon structures on anode performance. The properties and performance of the four materials are summarised in Table 2. The two best performing materials are the reduced aerogel and sand. These two materials have the highest and lowest surface area among the four materials and the smallest and largest pore diameters respectively. This stands as an important example showing that SSA and pore properties are not the only key parameters determining porous silicon's success as an anode material. Liang et al.63 reported that the morphology of the SBA-15 and diatom, which is an aggregation of isolated particles, adversely influences the cycle life. The route created a 3 nm passivation layer of silica over the surface of the porous silicon. The aggregation of particles is therefore not beneficial as ionic and electronic conduction is hindered through silica layers. Post cycling TEM results of the diatom, SBA-15 and sand silicon appear somewhat different to those reported by Shen et al.9 and Liu et al.,42 further indicating that upon cycling these samples, with passivated silica layers, perform differently.
4 Challenges and opportunities
(1) It is clear that the surface area and porosity do not completely govern the success of silicon as a LIB anode. For this reason, it is imperative that researchers publish detailed characterisation of material properties such as surface areas, pore volumes, pore size distributions, crystalline properties and particle morphology.(2) The evolution of the pore structure in porous silicon during lithiation has not been studied in great detail. With the ability to see which pore structures are beneficial to improved performance, an in situ or ex situ method of evaluating the pore structure vs. cycle life would significantly improve the understanding in this area. Such insight would give researchers the information necessary to use the versatile magnesiothermic reduction to provide these porous material properties on a bulk scale.
(3) Cycling parameters and other testing variables such as electrolyte additives81 and binders75,82 are well known to improve the capacity and cycle life of silicon anodes. Testing criteria for the fabrication of electrodes and cells should be reported in detail. Equally as these variables have shown significant improvements in non-porous materials their effect on porous materials is still to be quantified.
Like for like comparison between reports may never be possible due to experimental inconsistencies. However, for better comparison in future, researchers should strive to publish porous silicon performance vs. a silicon standard material.
5 Conclusion
Silicon has outstanding features as an anode material for LIBs, primarily its high specific and volumetric energy density. The major drawback is hinged upon the large volume change associated with lithiation causing the reversible cycling capacity to be poor. The properties of porous silicon have been shown to be advantageous in increasing the capacity and extending the cycling life. If performed effectively magnesiothermic reduction offers a relatively facile bulk synthesis route to porous silicon materials with morphological control over the silicon product.Synthesis via magnesiothermic reduction has been shown to offer the possibility of using templated silicon analogues from the vast catalogue of synthetic and natural sources. This method provides an instrument to study the varying effect of the porous silicon morphology on the anode performance. The reduction is a highly exothermic reaction and efforts must be made to avoid excessive rises in the reaction temperature. A number of studies have shown that slower ramping rates and thermal moderators can overcome excessive heat accumulation. Additionally, two step synthesis with lower enthalpies has been shown not only to avoid template destruction but provide oxide layers linked to improved cycling performance. The exothermic nature of the reaction means that the true reaction temperature can be well above the set conditions, and reported in excess of 1300 °C. It can therefore be difficult to strictly control and study the effect of temperature on the reaction. The reports of initiating the reaction at higher temperatures and then lowering the reaction temperature are interesting regarding both control and efficiency aspects.
The magnesiothermic reduction template assisted method leads to a mainly mesoporous silicon product with a similar shape to the initial template, although mesopores can be introduced into samples even if the templating structure is non-porous. To better understand the processes occurring under magnesiothermic reduction conditions studies should report in detail all the reaction criteria recommended above. Additionally, complete characterisation of both the reactant and product structures is needed to see how reaction conditions may affect different templates.
Key to assessing the effectiveness of porous silicon as an anode is understanding the evolution of the porous structure during lithiation. Methods such as TEM have started to reveal these behaviours.9,42,63,83 More analysis of porous silicon would be a valuable addition to understand how the pore structure evolves under cycling.
The high surface areas and pore volumes along with the nanocrystalline properties of magnesiothermically reduced silicon are advantageous for a number of additional applications in fields such as photoluminescence,31 solar power,32 photocatalysis,33 drug delivery,34 and catalysis support.35 Understanding the magnesiothermic reduction mechanism of producing porous silicon is crucial for the expansion of this technique. A clear design pathway should be identifiable from feedstock silica to desired silicon properties.
Conflicts of interest
There are no conflicts to declare.Abbreviations
| LIB | Lithium-ion battery |
| CVD | Chemical vapour deposition |
Acknowledgements
This work is supported by the EPSRC EP/L016818/1 and EP/R041822/1 grants.References
- M. Yoshio, R. Brodd and A. Kozawa, Lithium-Ion Batteries, Springer, 2009 Search PubMed.
- J. B. Goodenough and K. Park, J. Am. Chem. Soc., 2013, 135, 1167 CrossRef PubMed.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854 RSC.
- M. N. Obrovac, L. Christensen, D. B. Le and J. R. Dahn, J. Electrochem. Soc., 2007, 154, A849 CrossRef.
- M. N. Obrovac and L. J. Krause, J. Electrochem. Soc., 2007, 154, A103 CrossRef.
- J. Li and J. R. Dahn, J. Electrochem. Soc., 2007, 154, A156 CrossRef.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966 CrossRef PubMed.
- B. Key, R. Bhattacharyya, M. Morcrette, V. Seznec, J. M. Tarascon and C. P. Grey, J. Am. Chem. Soc., 2009, 131, 9239 CrossRef PubMed.
- C. Shen, M. Ge, L. Luo, X. Fang, Y. Liu, A. Zhang, J. Rong, C. Wang and C. Zhou, Sci. Rep., 2016, 6, 31334 CrossRef PubMed.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522 CrossRef PubMed.
- M. T. McDowell, S. W. Lee, J. T. Harris, B. A. Korgel, C. Wang, W. D. Nix and Y. Cui, Nano Lett., 2013, 13, 758 CrossRef PubMed.
- L. Y. Beaulieu, S. D. Beattie, T. D. Hatchard and J. R. Dahn, J. Electrochem. Soc., 2003, 150, A419 CrossRef.
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414 CrossRef.
- R. Ruffo, S. S. Hong, C. K. Chan, R. A. Huggins and Y. Cui, J. Phys. Chem. C, 2009, 113, 11390 CrossRef.
- N. Dimov, K. Fukuda, T. Umeno, S. Kugino and M. Yoshio, J. Power Sources, 2003, 114, 88 CrossRef.
- K. Persson, V. A. Sethuraman, L. J. Hardwick and Y. Hinuma, J. Phys. Chem. Lett., 2010, 1176 CrossRef.
- P. Yu, B. N. Popov, J. A. Ritter and R. E. White, J. Electrochem. Soc., 1999, 146, 8 CrossRef.
- M. D. Levi and D. Aurbach, J. Electroanal. Chem., 1997, 421, 79 CrossRef.
- M. Ge, J. Rong, X. Fang and C. Zhou, Nano Lett., 2012, 12, 2318 CrossRef PubMed.
- J. Christensen and J. Newman, J. Solid State Electrochem., 2006, 10, 293 CrossRef.
- X. Zhang, W. Shyy and A. Marie Sastry, J. Electrochem. Soc., 2007, 154, S21 CrossRef.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. Liu, L. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949 CrossRef PubMed.
- X. Li, M. Gu, S. Hu, R. Kennard, P. Yan, X. Chen, C. Wang, M. J. Sailor, J. Zhang and J. Liu, Nat. Commun., 2014, 5, 1 Search PubMed.
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56 RSC.
- M. Ge, X. Fang, J. Rong and C. Zhou, Nanotechnology, 2013, 24, 422001 CrossRef PubMed.
- X. Su, Q. Wu, J. Li, X. Xiao, A. Lott, W. Lu, B. W. Sheldon and J. Wu, Adv. Energy Mater., 2014, 4, 1300882 CrossRef.
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7, 19 CrossRef PubMed.
- N. Lebedeva, F. Di Persio and L. Boon-Brett, Eur. Comm., 2016 Search PubMed.
- J. O. Odden, G. Halvorsen and H. Rong, Silicon Chem. Sol. Ind., 2008, vol. 9, p. 16 Search PubMed.
- B. G. Gribov and K. V Zinov, Inorg. Mater., 2003, 39, 653 CrossRef.
- L. T. Canham, Appl. Phys. Lett., 1990, 57, 1046 CrossRef.
- K. K. Larbi, M. Barati and a. McLean, Can. Metall. Q., 2011, 50, 341 CrossRef.
- F. Dai, J. Zai, R. Yi, M. L. Gordin, H. Sohn, S. Chen and D. Wang, Nat. Commun., 2014, 5, 1 Search PubMed.
- M. Guo, X. Zou, H. Ren, F. Muhammad, C. Huang, S. Qiu and G. Zhu, Microporous Mesoporous Mater., 2011, 142, 194 CrossRef.
- J. Llorca, A. Casanovas, T. Trifonov, A. Rodríguez and R. Alcubilla, J. Catal., 2008, 255, 228 CrossRef.
- Z. Bao, M. R. Weatherspoon, S. Shian, Y. Cai, P. D. Graham, S. M. Allan, G. Ahmad, M. B. Dickerson, B. C. Church, Z. Kang, H. W. Abernathy, C. J. Summers, M. Liu and K. H. Sandhage, Nature, 2007, 446, 172 CrossRef PubMed.
- E. K. Richman, C. B. Kang, T. Brezesinski and S. H. Tolbert, Nano Lett., 2008, 8, 3075 CrossRef PubMed.
- R. A. Rapp, A. Ezis and G. J. Yurek, Metall. Trans., 1973, 4, 1283 CrossRef.
- H. Jia, P. Gao, J. Yang, J. Wang, Y. Nuli and Z. Yang, Adv. Energy Mater., 2011, 1, 1036 CrossRef.
- X. Liu, Y. Gao, R. Jin, H. Luo, P. Peng and Y. Liu, Nano Energy, 2014, 4, 31 CrossRef.
- S. Haouli, S. Boudebane, I. J. Slipper, S. Lemboub, P. Gębara and S. Mezrag, Phosphorus, Sulfur Silicon Relat. Elem., 2018, 193, 280 CrossRef.
- N. Liu, K. Huo, M. T. McDowell, J. Zhao and Y. Cui, Sci. Rep., 2013, 3, 1 Search PubMed.
- W. Luo, X. Wang, C. Meyers, N. Wannenmacher, W. Sirisaksoontorn, M. M. Lerner and X. Ji, Sci. Rep., 2013, 3, 2222 CrossRef PubMed.
- M. Barati, S. Sarder, A. McLean and R. Roy, J. Non-Cryst. Solids, 2011, 357, 18 CrossRef.
- D. Cho, M. Kim, J. Hwang, J. H. Park, Y. L. Joo and Y. Jeong, Nanoscale Res. Lett., 2015, 10, 424 CrossRef PubMed.
- H. Wu, N. Du, X. Shi and D. Yang, J. Power Sources, 2016, 331, 76 CrossRef.
- Y. Zhang and J. Huang, J. Mater. Chem., 2011, 21, 7161 RSC.
- S. C. Davis, Stress and Microstructural Evolution During Shape Preserving Silica Magnesiothermic Reduction, Georgia Institute of Technology, 2013 Search PubMed.
- R. F. Shepherd, P. Panda, Z. Bao, K. H. Sandhage, T. A. Hatton, J. A. Lewis and P. S. Doyle, Adv. Mater., 2008, 20, 4734 CrossRef.
- L. Shi, W. Wang, A. Wang, K. Yuan and Y. Yang, J. Alloys Compd., 2016, 661, 27 CrossRef.
- J. Xie, G. Wang, Y. Huo, S. Zhang, G. Cao and X. Zhao, Electrochim. Acta, 2014, 135, 94 CrossRef.
- Y. Lai, J. R. Thompson and M. Dasog, Chem.–Eur. J., 2018, 24, 7913 CrossRef PubMed.
- K. Chen, Z. Bao, A. Du, X. Zhu, G. Wu, J. Shen and B. Zhou, Microporous Mesoporous Mater., 2012, 149, 16 CrossRef.
- W. Chen, Z. L. Fan, A. Dhanabalan, C. H. Chen and C. L. Wang, J. Electrochem. Soc., 2011, 158, A1055 CrossRef.
- W. C. Cho, H. J. Kim, H. I. Lee, M. W. Seo, H. W. Ra, S. J. Yoon, T. Y. Mun, Y. K. Kim, J. H. Kim, B. H. Kim, J. W. Kook, C. Y. Yoo, J. G. Lee and J. W. Choi, Nano Lett., 2016, 16, 7261 CrossRef PubMed.
- L. Batchelor, A. Loni, L. T. Canham, M. Hasan and J. L. Coffer, Silicon, 2012, 4, 259 CrossRef.
- W. Wang, Z. Favors, R. Ionescu, R. Ye, H. H. Bay, M. Ozkan and C. S. Ozkan, Sci. Rep., 2015, 2, 2 Search PubMed.
- C. Li, C. Liu, W. Wang, Z. Mutlu, J. Bell, K. Ahmed, R. Ye, M. Ozkan and C. S. Ozkan, Sci. Rep., 2017, 1 Search PubMed.
- Z. Favors, W. Wang, H. H. Bay, Z. Mutlu, K. Ahmed, C. Liu, M. Ozkan and C. S. Ozkan, Sci. Rep., 2014, 4, 5623 CrossRef PubMed.
- M. Dasog, Z. Yang and J. G. C. Veinot, CrystEngComm, 2012, 14, 7576 RSC.
- S. Liu, B. Liu, Y. Yao, P. Dong and S. Zhao, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2016, 31, 965 CrossRef.
- A. Xing, S. Tian, H. Tang, D. Losic and Z. Bao, RSC Adv., 2013, 3, 10145 RSC.
- J. Liang, X. Li, Z. Hou, W. Zhang, Y. Zhu and Y. Qian, ACS Nano, 2016, 10, 2295 CrossRef PubMed.
- J. Liang, X. Li, Z. Hou, C. Guo, Y. Zhu and Y. Qian, Chem. Commun., 2015, 51, 7230 RSC.
- S. Sim, P. Oh, S. Park and J. Cho, Adv. Mater., 2013, 25, 4498 CrossRef PubMed.
- F. Lepoivre, D. Larcher and J. Tarascon, J. Electrochem. Soc., 2016, 163(13), A2791–A2796 CrossRef.
- S. Choi, T. Bok, J. Ryu, J. I. Lee, J. Cho and S. Park, Nano Energy, 2015, 12, 161 CrossRef.
- D. S. Jung, M. H. Ryou, Y. J. Sung, S. B. Park and J. W. Choi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12229–12234 CrossRef PubMed.
- C. Wang, J. Ren, H. Chen, Y. Zhang, K. Ostrikov, W. Zhang and Y. Li, Mater. Chem. Phys., 2015, 173, 89 CrossRef.
- Y. Yu, L. Gu, C. B. Zhu, S. Tsukimoto, P. A. van Aken and J. Maier, Adv. Mater., 2010, 22, 2247 CrossRef PubMed.
- K. H. Kim, D. J. Lee, K. M. Cho, S. J. Kim, J.-K. Park and H.-T. Jung, Sci. Rep., 2015, 5, 9014 CrossRef PubMed.
- S. Zhu, C. Zhu, Q. Meng, Z. Guo, Z. Yu, T. Lu, Y. Li, D. Zhang and W. M. Lau, RSC Adv., 2013, 6141 RSC.
- L. Wu, J. Yang, J. Tang, Y. Ren, Y. Nie and X. Zhou, Electrochim. Acta, 2016, 190, 628 CrossRef.
- A. Magasinski, B. Zdyrko, I. Kovalenko, B. Hertzberg, R. Burtovyy, C. F. Huebner, T. F. Fuller, I. Luzinov and G. Yushin, ACS Appl. Mater. Interfaces, 2010, 2, 3004 CrossRef PubMed.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Sci. 80, 2011, 334, 75 CrossRef PubMed.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. Fredrickson, B. Chmelka and G. Stucky, Sci. 80, 1998, 279, 548 CrossRef.
- H.-C. Tao, L.-Z. Fan and X. Qu, Electrochim. Acta, 2012, 71, 194 CrossRef.
- M. S. Wang, L. Z. Fan, M. Huang, J. Li and X. Qu, J. Power Sources, 2012, 219, 29 CrossRef.
- H. Kim, B. Han, J. Choo and J. Cho, Angew. Chem., Int. Ed., 2008, 47, 10151 CrossRef PubMed.
- L. Shen, X. Guo, X. Fang, Z. Wang and L. Chen, J. Power Sources, 2012, 213, 229 CrossRef.
- C. C. Nguyen and B. L. Lucht, J. Electrochem. Soc., 2014, 161, 1933 CrossRef.
- D. Mazouzi, Z. Karkar, C. R. Hernandez, P. J. Manero, D. Guyomard, L. Roué and B. Lestriez, J. Power Sources, 2015, 280, 533 CrossRef.
- D. Qiu, G. Bu, B. Zhao and Z. Lin, J. Solid State Electrochem., 2015, 19, 935 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta06370b |
| ‡ Present address: Faradion Limited, The Innovation Centre, 217 Portobello, Sheffield, S1 4DP. |
| This journal is © The Royal Society of Chemistry 2018 |