
Iodide-induced differential control of metal ion reduction rates: synthesis of terraced palladium–copper nanoparticles with dilute bimetallic surfaces†
 a
and
Michelle L.
Personick
*a
a
and
Michelle L.
Personick
*a
Abstract
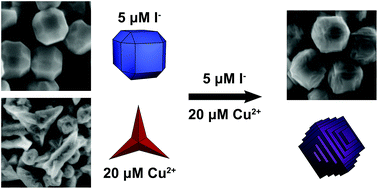
Metal nanoparticles possessing a high density of atomic steps and edge sites provide an increased population of undercoordinated surface atoms, which can enhance the catalytic activity of these materials compared to low-index faceted or bulk materials. Simply increasing reactivity, however, can lead to a concurrent increase in undesirable, non-selective side products. The incorporation of a second metal at these reactive stepped features provides an ideal avenue for finely attenuating reactivity to increase selectivity. A major challenge in synthesizing bimetallic nanomaterials with tunable surface features that are desirable for fundamental catalytic studies is a need to bridge differences in precursor reduction potentials and metal lattice parameters in structures containing both a noble metal and a non-noble metal. We report the use of low micromolar concentrations of iodide ions as a means of differentially controlling the relative reduction rates of a noble metal (palladium) and a non-noble metal (copper). The iodide in this system increases the rate of reduction of palladium ions while concurrently slowing the rate of copper ion reduction, thus providing a degree of control that is not achievable using most other reported means of tuning metal ion reduction rate. This differential control of metal ion reduction afforded by iodide ions enables access to nanoparticle growth conditions in which control of palladium nanoparticle growth by copper underpotential deposition becomes possible, leading to the generation of unique terraced bimetallic particles. Because of their bimetallic surface composition, these terraced nanoparticles exhibit increased selectivity to acetaldehyde in gas phase ethanol oxidation.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

