
On the role of hydroxide species in sulphur- and nitrogen-doped cobalt-based carbon catalysts for the oxygen evolution reaction†

Abstract
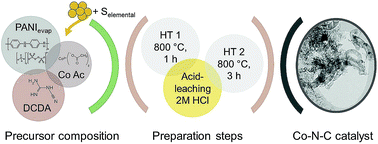
The influence of high S/Co ratios on the structural composition and oxygen evolution reaction (OER) activity of a group of cobalt-based carbon catalysts was investigated. Catalysts were prepared from polyaniline, cobalt acetate and dicyandiamide as precursors for active site formation and as structure forming agents. The sulphur to cobalt ratio was investigated in a range of S/Co = 10 to 32. On the basis of a comprehensive structural characterisation by XRD, Raman, XPS, TEM and N2 sorption measurements it was possible to show that the S/Co ratio has a significant impact on the carbon morphology. In fact, with increasing S/Co ratio the carbon morphology continuously changes from highly amorphous carbon to carbon-nanotubes, with increasing diameter. Besides the anticipated CoN4 sites and cobalt sulphite species, the catalysts also contained cobalt nanoparticles as well as cobalt hydroxide species. The most active catalyst required 0.37 ± 0.01 V overpotential to reach 10 mA cm−2 and even increased in activity during galvanostatic treatment and cycling-illustrating its very good performance. A faradaic efficiency of >35% was determined. A detailed analysis of the activity and stability in combination with Raman and XPS provides two explanations for observed Tafel slope changes, that might also be coupled to each other, namely a change in the carbon oxidation rate depending on preparation and potential or a variation in the coverage by hydroxide and oxidic species of the metal, whereas hydroxide species seem to enable a higher OER activity.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

