
Cs0.15FA0.85PbI3 perovskite solar cells for concentrator photovoltaic applications†
Nicola
Gasparini  a
and
Derya
Baran
*a
a
and
Derya
Baran
*a
a
and
Derya
Baran
*a
Abstract
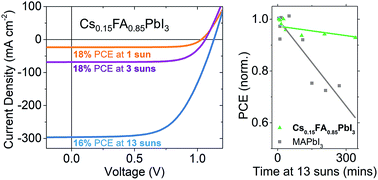
Recently developed, highly stable perovskite materials show promise for use in concentrator photovoltaics where the illumination intensity far exceeds standard test conditions. Here, we demonstrate solar cell devices employing different perovskite absorber layers featuring balanced charge generation and extraction characteristics at high light intensities greater than 10 suns. Using a mixed cesium-formamidinium perovskite, we are able to achieve over 18% PCE at 1 sun and 16% PCE at 13 suns with negligable performance loss after several hours of high intensity light soaking.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

