
Electrostatically regulated photoinduced electron transfer in “cationic” eco-friendly CuInS2/ZnS quantum dots in water†

Abstract
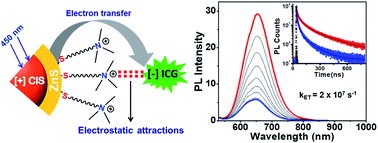
The potency of eco-friendly copper indium sulfide/zinc sulfide core/shell quantum dots (CIS/ZnS QDs) as efficient light harvesters in water is presented. A place exchange protocol is developed to prepare the much demanded cationic ([+]) CIS/ZnS QDs carrying a permanent positive charge, with ∼60% retention of the QD photoluminescence (PL) in water. Both steady-state and time-resolved photophysical studies confirm efficient electron transfer from the photoexcited CIS/ZnS QDs to indocyanine green (ICG) dye. The electrostatic attraction between the oppositely charged [+] CIS/ZnS QDs and [−] ICG dye is responsible for the formation of a strong ground state complex, which is vital for achieving an efficient electron transfer process in water. The successful demonstration of the efficient light harvesting properties using [+] CIS/ZnS QDs will be decisive in the development of artificial photosynthetic systems based on eco-friendly quantum dots.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

