
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(arylene alkylene)s with pendant N-spirocyclic quaternary ammonium cations for anion exchange membranes†
Thanh Huong
Pham
 a,
Joel S.
Olsson
a and
Patric
Jannasch
*ab
a,
Joel S.
Olsson
a and
Patric
Jannasch
*ab
aPolymer & Materials Chemistry, Department of Chemistry, Lund University, P.O. Box 124, SE-221 00 Lund, Sweden. E-mail: patric.jannasch@chem.lu.se
bUniversity of Tartu, Institute of Technology, Nooruse 1, EE-50411 Tartu, Estonia
First published on 1st August 2018
Abstract
Aromatic polymers functionalised with cycloaliphatic quaternary ammonium (QA) cations are currently emerging as base-stable anion exchange membranes (AEMs) for use in alkaline fuel cells and water electrolyzers. In the present work, we first prepared poly(biphenyl piperidine)s by superacid-mediated polycondensations, and then introduced different N-spirocyclic QA cations via cyclo-quaternisation of the piperidine rings. The resulting polymers and AEMs were free of diaryl ether linkages and benzylic C–H bonds, and showed very high thermal stability and hydroxide ion conductivity. Alkaline testing up to 120 °C implied that the alkaline stability of the spirocyclic cations was limited by distortions of the ring conformations caused by the rigid polymer backbone. As a consequence, the ring directly attached to the backbone degraded significantly faster by Hofmann β-elimination than the pendant ring in the spirocyclic cations. These results provide valuable insights towards the molecular design of highly thermochemically stable AEMs functionalised with N-spirocyclic QA cations.
Introduction
Fuel cells and water electrolysis cells are emerging as efficient and environmentally benign electrochemical energy conversion devices.1–10 These devices are typically operated under highly acidic conditions, utilizing proton exchange membranes such as Nafion®, and require expensive and rare platinum metal group catalysts.9–11 As a consequence, alkaline fuel cells and electrolysis cells operating with anion (hydroxide) exchange membranes (AEMs) are gaining considerable interest because the high pH conditions allow the use of catalysts based on non-noble metals, such as Fe, Co and Ni, as well as a wider choice of fuels.1–4 One of the major challenges in the development of these alkaline systems is to molecularly design and synthesize AEMs that combine high hydroxide ion conductivity with sufficient thermochemical stability over long periods of operation.Because of their highly basic and nucleophilic character, hydroxide ions may readily react with and degrade both the polymer backbone and quaternary ammonium (QA) cations of the AEM, resulting in severe loss of conductivity and mechanical integrity. Consequently, polymer backbones containing diaryl ether bridges have been found to be susceptible to cleavage, in particular if these bridges are activated by nearby electron-withdrawing groups.12–14 Depending on the structure, QA cations may degrade via a large number of different mechanisms including nucleophilic substitution at α-carbons, Hofmann β-elimination and different rearrangement reactions.15–21 In the intensive search for cations with high alkaline stability, certain heterocycloaliphatic QA cations15 and sterically hindered imidazolium cations22–24 have been reported to possess outstanding alkaline stability. For example, N,N-dimethylpiperidinium and 6-azonia-spiro[5.5]undecane-6-ium cations show approximately 21 and 26 times longer halftimes, respectively, than the conventional benzyltrimethyl QA cation in 6 M NaOH at 160 °C.15 The high stability of the former cations has been ascribed to low ring strain and to constrained conformations imposed by the ring structure, which increase the transition state energy of both substitution and elimination reactions.15 These results have inspired the preparation and study of a range of different AEMs functionalised with piperidinium25–29 and spirocyclic QA cations.13,30–35 For example, we recently prepared poly(arylene N,N-dimethylpiperidinium)s by polycondensation of N-methyl-4-piperidone and electron-rich phenyl monomers via superelectrophilic activation in triflic acid, followed by quaternisation reactions with iodomethane.26 AEMs based on these polymers showed no structural degradation after storage in 2 M NaOH at 60 °C after 360 h and a mere 5% ionic loss at 90 °C after the same period of time. In another approach, we prepared N-spirocyclic QA ionenes (spiro-ionenes) by cyclo-polycondensation of tetrakis(bromomethyl)benzene and dipiperidines.31 These polyelectrolytes showed excellent thermal and alkaline stability, with only slight degradation after 336 h in 1 M KOD in D2O at 120 °C.
In the current work, we have in two steps incorporated N-spirocyclic QA cations into poly(biphenylene alkylene)s which are free of any base-sensitive diaryl ether linkages and benzylic C–H. In the first step, precursor copolymers were prepared in superacid-mediated Friedel–Crafts type polycondensation of commercially available biphenyl, 4-piperidone and trifluoromethylketones (Scheme 1). In the next step, the secondary piperidine rings directly attached to the copolymer backbones were cyclo-quaternised using an α,ω-dibromoalkane to produce the N-spirocyclic QA cations. AEMs based on these copolymers were then prepared by solution casting and characterised regarding water uptake, hydroxide ion conductivity and thermochemical stability under alkaline conditions.
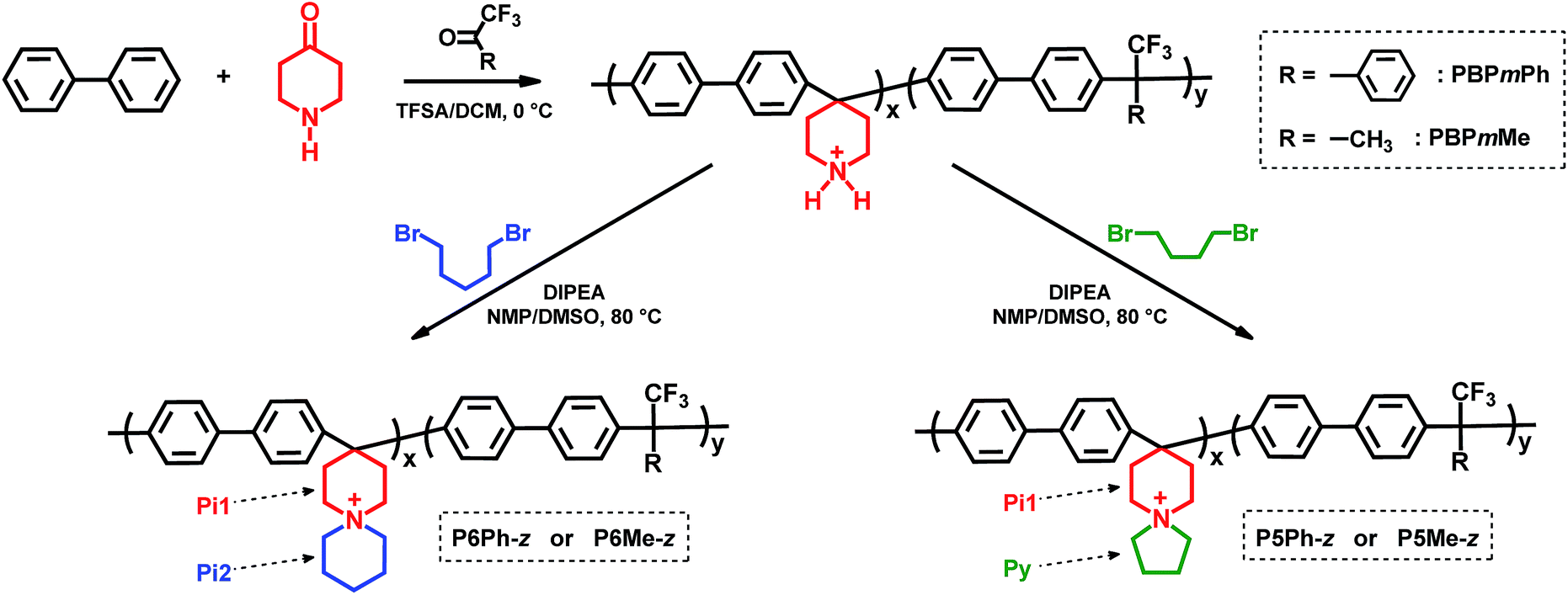 | ||
| Scheme 1 Synthesis pathway to poly(arylene alkylene)s functionalised with N-spirocyclic QAs via copolycondensation and cyclo-quaternisation (anions omitted for clarity). The dashed arrows indicate ring designations, and m and z denote the mol% of biphenyl piperidine units and the IEC value of the copolymers in the hydroxide form, respectively. | ||
Results and discussion
Polymer synthesis and characterisation
The precursor copolymers were prepared by Friedel–Crafts type copolycondensation of biphenyl, 4-piperidone and trifluoromethylketones, using trifluoromethanesulfonic acid (TFSA) as the catalyst (Scheme 1). Either 2,2,2-trifluoroacetophenone (TFAp) or 1,1,1-trifluoroacetone (TFAc) was used as the co-monomer in order to reduce the ionic content of the subsequent AEMs. Consequently, the ion exchange capacity (IEC, mequiv g−1) was controlled by adjusting the piperidone-to-trifluoromethylketone feed ratio. In addition, the monomer feed ratio and the type of co-monomer influenced the rigidity of the polymer backbone.Klumpp et al. have previously reported on the mechanism of the superacid-mediated polycondensation reaction of 4-piperidone and phenyl monomers in detail.36 In the first step, both the carbonyl and basic secondary amine groups of 4-piperidone are protonated by TFSA to form super electrophilic dications, which rapidly react with electron-rich biphenyl in the second step.36 In the case of TFAp and TFAc, only the ketone group is protonated. Still, the strongly electron-withdrawing trifluoromethyl group increases the electrophilicity of the cations significantly, thus enabling the subsequent condensation with biphenyl.37 Because the first step is rate-determining, the presence of a small excess of the ketone monomers dramatically enhances the polymerisation rate and the molecular weight of the resulting polymers.36,38 However, using too large excess of ketones may instead lead to crosslinking and gelation.38 Consequently, we used an excess of the ketones corresponding to 10–15 mol% of the biphenyl in the present copolycondensation. The resulting precursor copolymers containing TFAp and TFAc residues are denoted as PBPmPh and PBPmMe, respectively, where m indicates the mol% of biphenyl piperidine units, as calculated from 1H NMR spectra (Fig. 1a and d). To facilitate the 1H NMR analysis, trifluoroacetic acid (TFA) was added to the DMSO-d6 solutions in order to protonate the water residue in the solution, thus increasing its chemical shift to expose sample signals originally hidden by its broad signal. The copolymer compositions were calculated by comparing the signals originating from the Hα of the protonated secondary piperidinium rings at 3.1 ppm with the combined aromatic signals in the region of 7.0–8.0 ppm. Consistently, the content of biphenyl piperidine units in the copolymers was equal or lower than that predicted from the monomer feed, presumably because of the lower reactivity of the piperidone in relation to the trifluoromethylketones. Hence, in the preparation of PBP55Ph, PBP69Ph and PBP60Me, the starting mixture of 4-piperidone and trifluoromethylketone monomers contained 65, 75 and 60 mol%, respectively, of the former monomer.
 | ||
| Fig. 1 1H NMR spectra of (a) PBP69Ph, (b) P5Ph-2.3, (c) P6Ph-2.2, (d) PBP60Me, (e) P5Me-2.2 and (f) P6Me-2.0. TFA was added to shift the water signals (originally at ∼3.3 ppm) to above 10 ppm, revealing sample signals between 3.0 and 3.5 ppm. The signals between 7.0 and 8.0 ppm arose from the aromatic protons. | ||
In the neutral (non-protonated) state, the piperidine rings directly attached to the copolymer backbones (denoted as Pi1) in the precursor copolymers are strongly hydrogen bonding, leading to low solubility in common organic solvents such as dimethyl sulfoxide (DMSO), NMP (N-methyl-2-pyrrolidone), chloroform, and methanol. However, in the protonated state, the copolymers readily dissolved in DMSO, NMP and methanol, but remained insoluble in chloroform. Because of the limited solubility, we were unable to determine the molecular weight of the copolymers by size exclusion chromatography. Alternatively, the intrinsic viscosity ([η]) was determined to provide a measure of the molecular weight obtained. Thus, the intrinsic viscosity of the protonated PBP55Ph, PBP69Ph and PBP60Me copolymers was determined to be 0.17, 0.20 and 0.23 dL g−1, respectively, in DMSO at 25 °C, which suggested medium molecular weights.
In the second synthesis step, the Pi1 piperidine rings of the precursor copolymers were cyclo-quaternised using 1,4-dibromobutane and 1,5-dibromopentane to form pendant pyrrolidine (Py) and piperidine (Pi2) rings, respectively, by following our previously reported method (Scheme 1).13 The reaction was facilitated by the N,N-diisopropylethylamine (DIPEA) catalyst which converted the starting protonated secondary amine and intermediate tertiary amine moieties to their respective nucleophilic forms, enabling two successive nucleophilic (SN2) attacks to form the N-spirocyclic QA cations. To prevent precipitation of the neutralised copolymers, as well as side reactions that might cause crosslinking and gelation, DMSO solutions of the protonated precursor copolymers were added dropwise to solutions containing DIPEA and the respective dibromoalkane at 80 °C. The polymer concentration was kept low (approx. 3 wt%) throughout the reactions to promote the desired intramolecular cyclisation. In total, six different poly(biphenylene alkylene)s functionalised with N-spirocyclic QA cations were successfully synthesised. The copolymers are denoted as PyPh-z or PyMe-z, where Ph and Me indicate the use of TFAp and TFAc, respectively, y is the size of the pendant ring formed by cyclo-quaternisation (i.e., y = 5 and 6 for Py and Pi2, respectively), and z is the IEC value of the copolymer in the hydroxide form, calculated from Mohr titrations (Table 1).
| Polymer | IECBr (mequiv. g−1) | IECOHb (meqiv. g−1) | WUOH,80 °Cc (wt%) | λ OH,80 °C | SWOH,80 °Cc in-plane (%) | SWOH,80 °Cc through-plane (%) | σ OH,80 °Cc (mS cm−1) | T d,95 (°C) | |
|---|---|---|---|---|---|---|---|---|---|
| NMRa | Titrated | ||||||||
| a Calculated from the 1H NMR spectra of precursor polymers. b Calculated from titrated IECBr values. c Measured at 80 °C under fully hydrated conditions (immersed). d Measured by TGA under N2 at 10 °C min−1. e AEM was too brittle and could not be analyzed. | |||||||||
| P5Ph-1.8 | 1.60 | 1.63 | 1.82 | 76 | 23 | 20 | 25 | 65 | 365 |
| P6Ph-1.8 | 1.57 | 1.63 | 1.81 | NAe | NAe | NAe | NAe | NAe | 344 |
| P5Ph-2.3 | 1.97 | 2.00 | 2.29 | 187 | 45 | 39 | 31 | 102 | 368 |
| P6Ph-2.2 | 1.91 | 1.94 | 2.20 | 220 | 55 | 49 | 22 | 94 | 352 |
| P5Me-2.2 | 1.88 | 1.94 | 2.21 | 96 | 24 | 25 | 40 | 95 | 362 |
| P6Me-2.0 | 1.83 | 1.81 | 2.05 | 97 | 26 | 21 | 46 | 102 | 337 |
The molecular structure of the cationic copolymers was confirmed by 1H NMR spectroscopy (Fig. 1 and S1†). After cyclo-quaternisation, signal c, originating from protonated amine protons in precursor polymers, totally disappeared. The shifts of signal a, originating from the Hβ of the Pi1 rings, increased from ∼2.6 to ∼2.7 ppm. Shifts from the Hα of Pi1 (b) also increased from ∼3.0 to 3.4–3.5 ppm (Fig. 1 and S1†). In the case of P5Ph-z and P5Me-z, new signals emerged at ∼3.3 and ∼2.0 ppm, which were assigned to the Hα and Hβ of the newly formed Py rings (Fig. 1b and e, and S1b†). For P6Ph-z and P6Me-z, the formation of Pi2 rings was confirmed by the appearance of signals at 3.3, 1.7 and 1.5 ppm, assigned to the Hα, Hβ and Hγ, respectively, of these rings (Fig. 1c and f, and S1c†).
Membrane preparation
AEMs based on the copolymers were prepared by casting from 5 wt% solutions of the copolymers in DMSO at 85 °C. All copolymers, except P6Ph-1.8, formed mechanically tough, transparent and flexible membranes (Fig. 2). The brittleness of P6Ph-1.8 was probably the result of its medium molecular weight combined with high polymer chain rigidity. All AEMs were exchanged to the bromide form by immersion in 1 M aq. NaBr solution at 50 °C prior to analysis. As seen in Table 1, the IEC values determined by Mohr titration of the membranes were in good agreement with the expected IECs calculated from the 1H NMR spectra of the precursor polymers, confirming the high efficiency of the cyclo-quaternisation reactions. Small angle X-ray scattering (SAXS) was employed to study the segregation of the ions in the AEMs in the bromide form at 75% RH. The resulting SAXS profiles indicated no distinct ionomer peak (Fig. S2†), most probably because the high rigidity of both the N-spirocyclic QAs and the polymer backbone prevented efficient ion clustering. This finding is consistent with our previous results on poly(arylene piperidinium) AEMs, where poly(arylene N,N-dimethylpiperidinium)s were found not to show any distinct ionomer peak.26 | ||
| Fig. 2 Photographs of (a) PBP69Ph and (b) P6Ph-2.2, showing their transparency and foldability as AEMs. | ||
Water uptake and swelling
Sufficient water uptake (WU) is crucial for the formation of a percolating system of water-rich channels required for transportation of anions across the AEM.39 However, a too high water uptake will dilute the ion (charge carrier) concentration in the membrane, which decreases the anion conductivity. Furthermore, high water contents lead to excessive swelling and deteriorate the mechanical properties of the AEM. In the present work, membrane P5Ph-1.8 with the lowest IEC value reached the lowest water uptake, 76 wt% at 80 °C (Fig. 3a). This membrane also had the lowest swelling (SW) at 80 °C in both in-plane and through-plane, i.e., 20 and 25%, respectively (Table 1). Membranes P5Ph-2.3 and P6Ph-2.2 showed the highest water uptake, increasing from 90 and 100 wt%, respectively, at 20 °C to 190 and 220 wt%, respectively, at 80 °C. These AEMs also reached the highest in-plane swelling of 39 and 49%, respectively, at 80 °C. In comparison, membranes P5Me-2.2 and P6Me-2.0 showed much more reasonable water uptake values: 55 and 50 wt%, respectively, at 20 °C and 96 and 97 wt%, respectively, at 80 °C (Fig. 3a). Even though they showed a high through-plane swelling of 40 and 46% at 80 °C, their in-plane swelling ratio at the same temperature was much lower, reaching only 25 and 21%, respectively. The reason for the much higher water uptake of P5Ph-2.3 and P6Ph-2.2, as well as the brittleness of P6Ph-1.8, is most likely due to the lower molecular weight of the PBPmPh precursor polymers in comparison with the PBPmMe ones, as indicated by the intrinsic viscosities.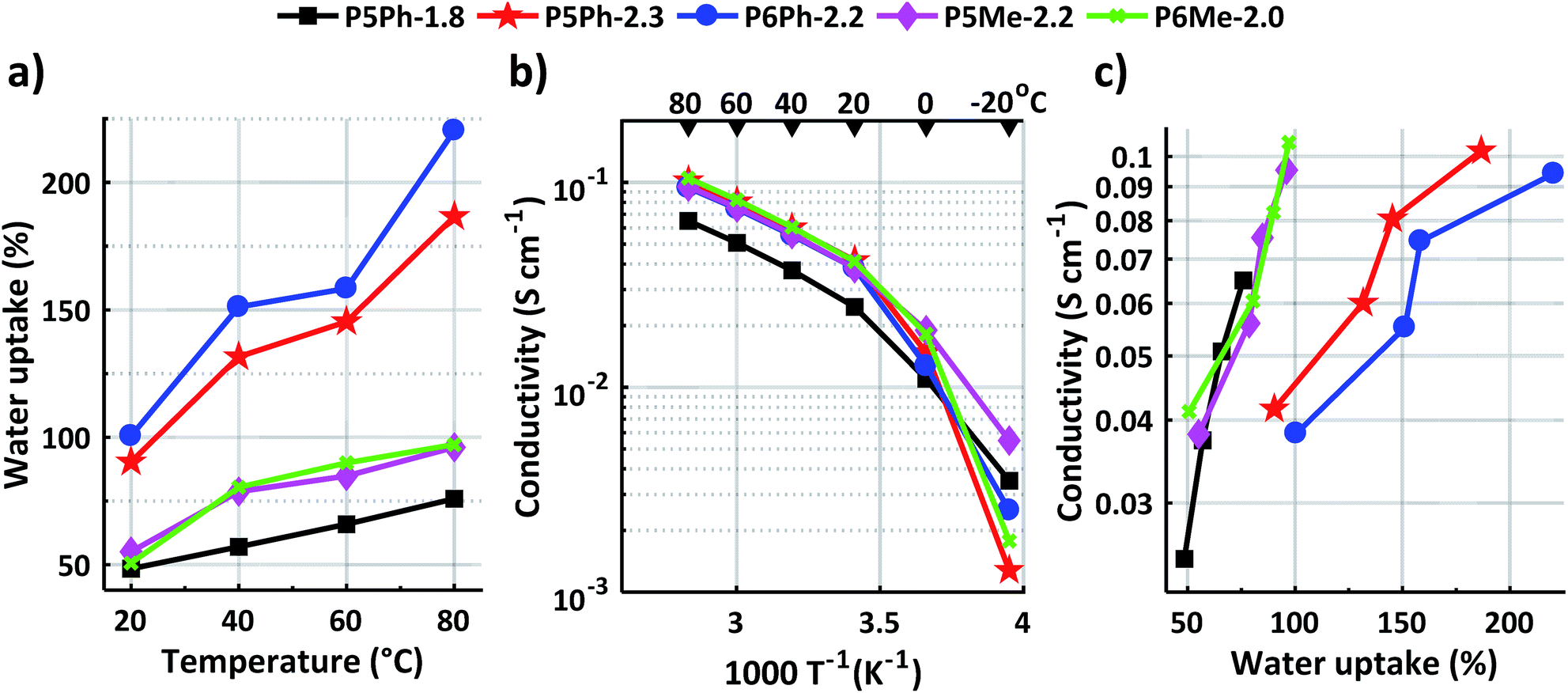 | ||
| Fig. 3 (a) Water uptake of fully hydrated AEMs in the hydroxide form as a function of temperature, (b) hydroxide conductivity of fully hydrated AEMs as a function of T−1, and (c) hydroxide conductivity of fully hydrated AEMs as a function of water uptake. | ||
Hydroxide conductivity
The temperature dependent hydroxide conductivity of fully hydrated AEMs was measured in the plane of the membranes by electrochemical impedance spectroscopy using a two probe-cell. During the measurement, the membranes were immersed in water in an enclosed cell while the temperature was varied between −20 and 80 °C. As seen in Fig. 3b, their hydroxide conductivity increased sharply up to 20 °C as a result of ice melting. Between 20 and 80 °C, the hydroxide conductivity showed an Arrhenius-like behaviour. As expected, membrane P5Ph-1.8 had the lowest conductivity, 65 mS cm−1 at 80 °C, due to its low IEC and water uptake. The four other AEMs of this study, P5Ph-2.3, P6Ph-2.2, P5Me-2.2, and P6Me-2.0, showed high and quite similar levels of hydroxide conductivity, despite the differences in their water uptake. At 80 °C, these membranes reached 102, 94, 95 and 105 mS cm−1, respectively. Between 20 and 80 °C, their conductivity showed an Arrhenius behaviour, with an estimated apparent activation energy Ea = 13–14 kJ mol−1. This is comparable to Ea values reported by Pan et al. for benzyltrimethylammonium-modified polysulfone membranes (13 kJ mol−1)40 and by our group for poly(phenylene oxide)s functionalised with monocyclic quaternary piperidinium cations (12–14 kJ mol−1).25Fig. 3c shows the relationship between the hydroxide conductivity and water uptake between 20 and 80 °C. As seen, the increase of the conductivity with the water uptake was much steeper for P5Ph-1.8, P5Me-2.2 and P6Me-2.0, than for P5Ph-2.3 and P6Ph-2.2. This finding suggested that the conductivity of the latter two AEMs was restricted by dilution effects resulting from their high water uptake. Hence, membranes P5Me-2.2 and P6Me-2.0 displayed the best balance of water uptake and conductivity in this study. Potentially, the size of the N-spirocyclic QA cation may affect both the water uptake and conductivity of the AEM. A comparison of membranes P6Me-2.0 and P5Me-2.2, containing the 6-membered Pi2 rings and the 5-membered Py rings, respectively, shows a quite similar water uptake and conductivity, although the latter AEM had a higher IEC. Thus, more data are required in order to assess the role of the ring size in this kind of AEM. The hydroxide conductivity of the present membranes may be compared with values reported by Bae's group for poly(biphenyl alkylene) membranes with QA cations placed on flexible pentyl spacers, which reached 88 mS cm−1 at 80 °C (IEC = 1.9 mequiv g−1, WU = 110 wt%).41 Moreover, we have previously reported a hydroxide conductivity of 111 mS cm−1 for an AEM based on poly(biphenyl N-hexyl-N-methylpiperidinium) at 80 °C (IEC = 2.1 mequiv g−1, WU = 170 wt%).26 Consequently, the conductivity of, e.g., membrane P6Me-2.0 in the present study, compared quite favourably with a conductivity of 105 mS cm−1 at 80 °C (IEC = 2.0 mequiv g−1, WU = 97 wt%).
Thermal stability
Long-term thermal and chemical stabilities under alkaline conditions are crucial properties that directly affect the lifetime of AEMs and electrochemical devices containing them. The thermal stability of both the protonated precursor polymers in the triflate form and the AEMs in the bromide form was studied by TGA under a N2 atmosphere, and the results in terms of thermal decomposition temperature (Td,95) values are presented in Table 1 (TGA traces in Fig. 4). The precursor polymers PBP55Ph, PBP69Ph and PBP60Me showed remarkably high Td,95 values in a quite narrow temperature range: 391, 387 and 383 °C, respectively (Fig. 4a). As expected, the Td,95 values of the AEMs were lower than those of the precursors but were still well above 300 °C (Fig. 4b). This exceptional thermal stability can be attributed to the high aromaticity and rigidity of the present cationic copolymers. The AEMs functionalised with Py rings reached Td,95 values in the narrow range between 362 and 368 °C. Hence, there was no significant difference in thermal stability between the copolymers containing TFAp and TFAc residues, respectively. Notably, the thermal stability of the present copolymers was significantly higher than that of poly(arylene piperidinium)s (Td,95 = 232–312 °C) with similar backbones, but functionalised with monocyclic QAs instead of spirocyclic ones.26 Obviously, the spirocyclic arrangement of the cations enhanced the thermal stability in relation to the monocyclic ones. The stability was also strongly influenced by the size of the N-spirocyclic QA cation. With a given precursor copolymer, the AEMs with Py rings consistently had a significantly higher Td,95 (by 16–25 °C) than those with Pi2 rings.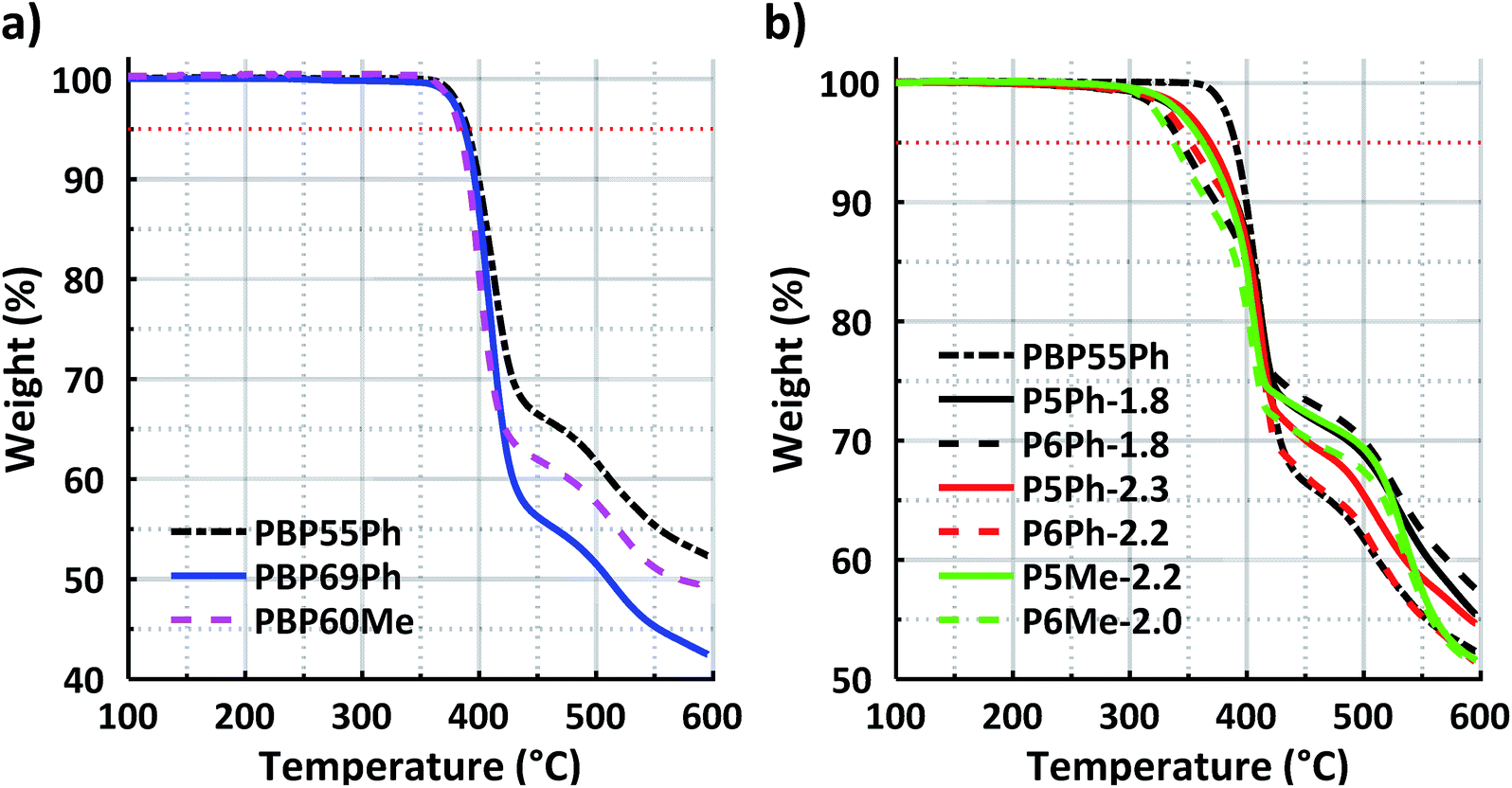 | ||
| Fig. 4 TGA profiles of (a) the protonated precursor polymers in the triflate form and (b) AEMs in the bromide form. The red dotted lines mark the 95% threshold. | ||
Alkaline stability
The alkaline stability of the AEMs was investigated by 1H NMR spectroscopy after storage in 2 M aq. NaOH at 90 and 120 °C during different periods of time. After the storage, the AEMs were ion-exchanged to the bromide form and dissolved in a DMSO-d6/TFA mixture for 1H NMR analysis. The 1H NMR spectra of P5Ph-1.8 and P6Ph-1.8 before and after treatment in NaOH at 90 °C are displayed in Fig. 5a and b, respectively (corresponding 1H NMR spectra of the other AEMs are shown in Fig. S3 and S4†). As seen, the AEMs showed signs of degradation indicated by the appearance of new signals (marked by red arrows). These new signals were barely detectable after 168 h, but increased over time to become apparent after 336 and 720 h. Furthermore, membranes P5Ph-1.8, P5Ph-2.3 and P5Me-2.2, bearing the 5-membered Py ring, became insoluble in both DMSO and DMSO/TFA after alkaline treatment for 720, 336 and 720 h, respectively. In contrast, P6Ph-1.8, P6Ph-2.2 and P6Me-2.0, containing the 6-membered Pi2 ring, remained soluble in the same solvents after the alkaline storage. This finding may suggest a difference in the degradation mechanism between the two N-spirocyclic QA cations. | ||
| Fig. 5 1H NMR spectra of (a) P5Ph-1.8 and (b) P6Ph-1.8 recorded in DMSO-d6/TFA before and after immersion in 2 M aq. NaOH at 90 °C for different periods of time. TFA was added to shift the water signals (originally at ∼3.3 ppm) to above 10 ppm, revealing sample signals between 3.0 and 3.5 ppm. The red arrows mark new signals originating from degradation products. Expansion of the regions between 4.7–6.7 and 8.7–10.4 ppm (marked by red dashed boxes) is shown for (c) P5Ph-1.8 after 336 h immersion and (d) P6Ph-1.8 after 720 h immersion. Possible products from Hofmann β-elimination are displayed in black dashed boxes for the two N-spirocyclic QA cations. | ||
Signals from degradation products were observed in the regions between 4.9 and 6.5, as well as between 9.3 and 10.0 ppm, which implied that the cations degraded via Hofmann β-elimination. Expansions of these regions of the 1H NMR spectra of P5Ph-1.8 after 336 h immersion and P6Ph-1.8 after 672 h storage are shown in Fig. 5c and d, respectively. The three signals at 6.5 (b), 5.3 (a1) and 4.9 ppm (a2) had intensity ratios of approx. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. This was in excellent agreement with the formation of alkene (
:1:1. This was in excellent agreement with the formation of alkene (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) and alkenyl (−CHCH2) protons, expected after ring-opening β-elimination at Pi1 to form the degradation products 1A and 1B shown in Fig. 5c and d, respectively. Moreover, the corresponding vinyl signals expected after ring-opening β-elimination at Pi2 and Py, to form the degradation products 2A and 2B, respectively, appeared as two signals at 5.7 (e) and 5.0 ppm (d). Because of restrictions imposed by the rigid polymer backbone, the rotation of the vinyl group around the quaternary carbon is limited in structures 1A and 1B. This explains the strong splitting of the alkene (CH2) protons. In addition, the electron-withdrawing aromatic rings in the backbone shift the signals downfield, giving them higher chemical shifts than the corresponding 1H NMR signals from 2A and 2B. The addition of TFA also generated signals c and f above 9.0 ppm, which corresponded to the (protonated) tertiary amines (R3−NH+) formed by degradation of the cations (Fig. 5c and d).
CH2) and alkenyl (−CHCH2) protons, expected after ring-opening β-elimination at Pi1 to form the degradation products 1A and 1B shown in Fig. 5c and d, respectively. Moreover, the corresponding vinyl signals expected after ring-opening β-elimination at Pi2 and Py, to form the degradation products 2A and 2B, respectively, appeared as two signals at 5.7 (e) and 5.0 ppm (d). Because of restrictions imposed by the rigid polymer backbone, the rotation of the vinyl group around the quaternary carbon is limited in structures 1A and 1B. This explains the strong splitting of the alkene (CH2) protons. In addition, the electron-withdrawing aromatic rings in the backbone shift the signals downfield, giving them higher chemical shifts than the corresponding 1H NMR signals from 2A and 2B. The addition of TFA also generated signals c and f above 9.0 ppm, which corresponded to the (protonated) tertiary amines (R3−NH+) formed by degradation of the cations (Fig. 5c and d).
The cationic loss in the AEMs occurring by β-elimination reactions was estimated by comparing the intensity of signals b and e with that of the combined aromatic signals (Fig. 5). Based on the intensity ratio between signals e and b, the degradation of the Pi2 and Py rings was significantly lower than that of the Pi1 ring, suggesting that β-elimination was more prominent in the restricted Pi1 than the presumably more relaxed Pi2 and Py rings. Due to the insolubility after the alkaline stability test, the cationic loss in P5Ph-1.8, P5Ph-2.3 and P5Me-2.2 could not be evaluated. After 720 h at 90 °C, the cationic loss via Hofmann elimination in the Pi1 rings of P6Ph-1.8, P6Ph-2.2 and P6Me-2.0 was estimated to be 23, 18 and 24%, respectively. The cationic loss via Hofmann elimination in the Pi2 ring was difficult to evaluate because signals d and e were nearly undetectable. This motivated us to perform an additional stability test at 120 °C to further confirm and quantify the presence of the degradation products (Fig. 6 and S5†). After 336 h in 2 M aq. NaOH at 120 °C, the cationic loss in membranes P6Ph-1.8, P6Ph-2.2 and P6Me-2.0 via Hofmann β-elimination in the Pi1 ring was estimated to be 50, 45 and 45%, respectively. At the same time, the degree of Hofmann β-elimination in the Pi2 ring was only 10, 8 and 10%, respectively.
 | ||
| Fig. 6 1H NMR spectra of P6Ph-1.8 recorded in DMSO-d6/TFA before and after immersion in 2 M aq. NaOH at 120 °C for 360 h. TFA was added to shift the water signals (originally at ∼3.3 ppm) to above 10 ppm, exposing sample signals between 3.0 and 3.5 ppm. The red arrows mark new signals originating from degradation products. | ||
The explanation for the reduced alkaline stability of the N-spirocyclic QA cations in the present copolymers, in comparison with the corresponding low molecular weight model substances,15 is most probably the distortion of the piperidinium ring conformation by the rigid copolymer backbone. According to Kreuer et al., the exceptional stability of the 6-azaspiro[5.5]undecan-6-ium cation originates from the low ring strain and restricted conformation by the spirane ring structure, which raises the energy level of the transition state in comparison with the starting piperidinium moiety.15 In the present AEMs, the Pi1 rings were directly attached to the bulky and stiff polymer backbone. This may significantly distort the bond angles and prevent conformational relaxation within the N-spirocyclic QA cations, thus reducing the activation energy and facilitating the degradation reactions. For this reason, the Pi1 rings were significantly more susceptible to ring-opening β-elimination than the pendant and apparently more relaxed Pi2 and Py rings. Especially in the AEMs that contained N-spirocyclic cations with exclusively 6-membered rings, i.e., P6Ph-1.8, P6Ph-2.2 and P6Me-2.0, this might be the only plausible explanation for the difference in the degradation rate of the Pi1 and Pi2 rings. This finding is in good agreement with our earlier results on spiro-ionenes.31 The introduction of a short alkyl spacer that increases flexibility and allows ring relaxation significantly increased the alkaline stability of the spiro-ionenes. The sample with the spacer showed no sign of Hofmann degradation after 336 h immersion in 1 M KOD/D2O at 120 °C. In contrast, the spiro-ionene without spacer still degraded through Hofmann elimination under the same conditions.31 Hence, distortion of ring conformations might severely reduce the advantage of the spirocyclic arrangement in comparison with a monocyclic one. Actually, the alkaline stability of the present N-spirocyclic QA cations proved to be lower than that of monocyclic N,N-dimethylpiperidinium cations of poly(arylene piperidinium)s, which showed less than 10% cationic loss after 720 h storage in 2 M NaOH at 90 °C.26
Besides β-elimination, the AEMs most probably also degraded by additional degradation mechanisms, e.g., via ring-opening nucleophilic substitutions (Scheme S1†). Unfortunately, these degradation pathways gave rise to 1H NMR signals that were overlapped with the alkylene signals from the original copolymers, as well as the signals from the degradation products from the Hofmann β-elimination, and were thus impossible to identify and quantify. Membranes P5Ph-1.8, P5Ph-2.3 and P5Me-2.2, bearing the 5-membered rings, might be more prone to these degradation pathways.15
Experimental
Materials
Biphenyl (99%, Acros), 2,2,2-trifluoroacetophenone (TFAp, 99%, Sigma-Aldrich), 1,1,1-trifluoroacetone (TFAc, 99%, Sigma-Aldrich), 1,1,1-trifluoroacetic acid (TFA, 99%, Acros), triflic acid (TFSA, 99%, Acros), 1,4-dibromobutane (99%, Acros), 1,5-dibromopentane (97%, Aldrich), N,N-diisopropylethylamine (DIPEA, ≥ 99%, Sigma Aldrich), isopropanol (IPA, reagent grade, VWR), diethyl ether (reagent grade, VWR), N-methyl-2-pyrrolidone (NMP, reagent grade, Acros), dimethyl sulfoxide (DMSO, reagent grade, VWR), NaBr (99%, Sigma-Aldrich), ethanol (99.5%, Solveco), NaOH (99% pellets, VWR), and DMSO-d6 (99,96 atom% D, Sigma-Aldrich) were all used as received. 4-Piperidone monohydrate hydrochloride (98%, Sigma-Aldrich) was washed with ethanol and dried under vacuum at room temperature before use. Dichloromethane was dried using an MBraun dry solvent dispenser system MB-SPS 800.Polymerisation
The precursor copolymers PBP55Ph, PBP69Ph and PBP60Me were prepared by Friedel–Crafts type polycondensation of biphenyl, 4-piperidone monohydrate hydrochloride, and either TFAp or TFAc, at 0 °C in anhydrous DCM using TFSA as the superacid catalyst. The IEC of the copolymers was controlled by adjusting the molar ratio between 4-piperidone monohydrate hydrochloride and TFAp or TFAc. The amounts of reagents and reactants, as well as the reaction times, are summarised in Table 2. Here follows a typical procedure exemplified by the synthesis of PBP60Me. To a 250 ml one-neck round flask was added biphenyl (3.80 g, 1 eq.), 4-piperidone monohydrate hydrochloride (2.50 g, 0.66 eq.), TFAc (0.97 ml, 0.44 eq.) and anhydrous DCM (10 ml). The reaction mixture was cooled to 0 °C using an ice bath before adding TFSA (22 ml, 10 eq.) dropwise using a funnel. The reaction mixture was then stirred at 0 °C using a magnetic stirrer for 8 h. The highly viscous reaction mixture was poured into iced water, resulting in the formation of large, hard polymer lumps. These lumps were crushed using a mortar and pestle, and then washed thoroughly with deionised water and dried under vacuum at room temperature for 24 h. Transparent, flexible films were cast from 5 wt% DMSO solutions of the different precursor polymers at 85 °C (Fig. 2a). These films were used in the subsequent synthesis and analysis.| Polymer | m biphenyl (g) | m 4-piperidone (g) | V TFAc (ml) | V TFAp (ml) | V DCM (ml) | V TFSA (ml) | Reaction time (h) |
|---|---|---|---|---|---|---|---|
| PBP55Ph | 2.03 | 1.50 | — | 0.75 | 4.5 | 13.4 | 4 |
| PBP69Ph | 1.75 | 1.50 | — | 0.46 | 4.5 | 11.5 | 3 |
| PBP60Me | 3.80 | 2.50 | 0.97 | — | 10 | 22 | 8 |
Cyclo-quaternisation
The precursor polymers were functionalised with pendant N-spirocyclic QA cations via cyclo-quaternisation using 1,4-dibromobutane and 1,5-dibromopentane (1.15 eq.), respectively, to obtain a series of six different cationic polymers. Here, the synthesis of P6Me-2.0 is described as an example. To a 50 ml two-neck round flask equipped with a condenser was added 1,5-dibromopentane (0.34 ml, 1.15 eq.), DIPEA (1.9 ml, 5 eq.), NMP (12 ml) and DMSO (12 ml). After heating the mixture to 80 °C, a solution of PBP60Me (1.19 g, 1 eq.) in DMSO (12 ml) at 80 °C was added dropwise under vigorous stirring. After 16 h, the reaction mixture was cooled to room temperature, and the product was precipitated in a mixture of diethyl ether and DCM, before being washed with diethyl ether, and then with 1 M aq. NaBr solution and finally with water. The polymer was then dried under vacuum at room temperature to give a pink/orange powder.Polymer characterisation
The molecular structure of the polymers was confirmed by 1H NMR spectra recorded at 400 MHz, using a Bruker DRX 400 spectrometer. The solutions used were based on either DMSO-d6 or a mixture of DMSO-d6 and TFA.The intrinsic viscosity of the precursor polymers PBP55Ph, PBP69Ph and PBP60Me was measured at 25 °C using an Ubbelohde viscometer. The samples were dried at 50 °C under vacuum for 48 h, weighed, and dissolved in 15 ml 0.1 M LiBr in DMSO solution (blank solution). These stock solutions were later diluted by adding the blank solution to reduce their concentrations (Cs). The resulting solutions were used immediately after preparation. The flow times through the capillary of the blank solution (tblank) and of the polymer solutions (tsample) were taken as the average of four measurements. The inherent (ηinh) and reduced (ηred) viscosities at four different concentrations were calculated as42
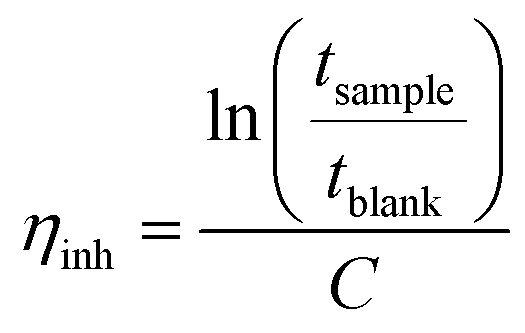 | (1) |
 | (2) |
The intrinsic viscosity ([η]) was calculated as the average of the intersections of the linear regressions of ηinh and ηred with the y-axis.
Membrane preparation
AEMs were prepared by casting 5 wt% solutions of the cationic polymers in DMSO. The general procedure is described here. Approximately 0.15 g polymer was dissolved in DMSO to obtain a 3 g solution. This solution was filtrated through a syringe-driven filter unit (ø = 25 mm, Fluoropore membrane, 5 μm) into a Petri dish (ø = 50 mm) which was then placed in a ventilated casting oven at 85 °C for at least 24 h. Afterward, the resulting AEM was immersed in 1 M aq. NaBr solution for at least 72 h to exchange all the remaining triflate ions to bromide ones. The AEM in the bromide form was transformed into the hydroxide form by ion-exchange in 1 M aq. NaOH solution for at least 48 h. This membrane was then thoroughly washed with degassed deionised water and stored in degassed deionised water under a nitrogen atmosphere.Thermal decomposition
The thermal decomposition of the AEMs in the bromide form and of precursor polymers in the protonated state with triflate counter ions was studied by thermogravimetric analysis (TGA) using a TGA Q500 (TA Instruments) during heating from 50 to 600 °C at 10 °C min−1 under a nitrogen atmosphere. In order to remove water residues, the samples were kept isothermally at 120 °C for 20 min prior to the analysis. The thermal decomposition temperature was reported at 5% weight loss (Td,95).Ion exchange capacity
The IEC of the AEMs in the bromide (IECBr) form was determined by titration. The membranes were first dried at 50 °C under vacuum during 48 h and weighed to obtain their dry weights. The dry membranes were then immersed in 25 ml 2 M aq. NaNO3 solution for 7 days. The solutions were titrated with an aq. AgNO3 solution (approx. 0.01 M), using an aq. K2CrO4 solution (0.1 M) as the indicator. The IEC of the AEMs in the hydroxide form (IECOH) was calculated from the IECBr as | (3) |
Water uptake and swelling
The water uptake and swelling of the AEMs in the hydroxide form were determined after equilibration at 20, 40, 60 and 80 °C, respectively. The membranes in the bromide form were dried at 50 °C under vacuum for 48 h, and then weighed and measured to obtain their dry weight (mdry,Br), length (ldry) and thickness (ddry). These membranes were then ion-exchanged to the hydroxide form. By assuming that all Br− was exchanged to OH−, the dry weight of the AEMs in the hydroxide form (mdry) was calculated from mdry,Br as| mdry = mdry,Br × (1 − 0.0629 × IECBr) | (4) |
The AEMs in the hydroxide form were immersed in deionised, degassed water at 20 and 40 °C for 24 h, and at 60 and 80 °C for 6 h to reach equilibrium. Next, the samples were wiped dry with tissue paper and quickly weighed and measured to obtain their weight (mwet), length (lwet) and thickness (dwet) in the hydrated state. Their water uptake was calculated as
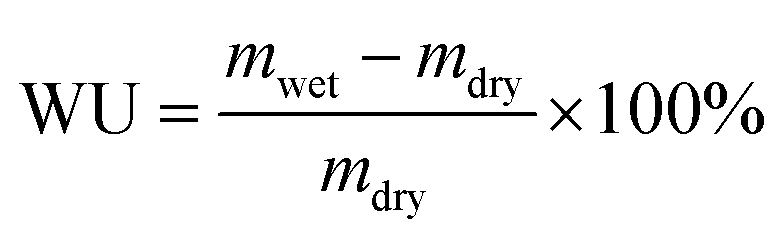 | (5) |
Assuming that in the dry state, the length and thickness of the membrane in the hydroxide form are similar to those in the bromide form, the in-plane and through-plane swelling (SW) was calculated as
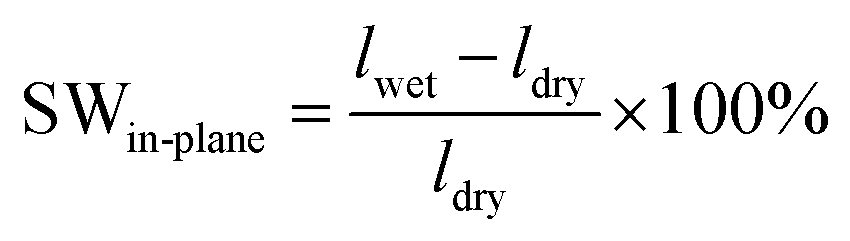 | (6) |
 | (7) |
Hydroxide conductivity
The hydroxide ion conductivity of the AEMs in the hydrated state was measured by electrochemical impedance spectroscopy using a two-probe set-up and a Novocontrol high-resolution dielectric analyzer V 1.01S. During the measurements, these membranes were kept hydrated in a closed cell filled with deionised degassed water. The voltage amplitude was kept at 50 mV while varying the frequency from 107 to 10−1 Hz in the temperature range of −20 to 80 °C.Small angle X-ray scattering
Phase separation by ionic clustering was studied by small angle X-ray scattering (SAXS) measurements of the AEMs in the bromide form at 75% RH. Pieces of the P6Ph-1.8, P6Ph-2.2 and P6Me-2.0 membranes were first equilibrated at 75% RH by enclosing them in a desiccator containing a saturated aq. NaCl solution for 7 days. These membranes were then transferred into SAXS cells which were sealed and kept in the desiccator at 75% RH prior to the measurements. Data were collected in the q-range of 0.14–7.5 nm−1 using a SAXSLAB SAXS instrument (JJ X-ray Systems Aps, Denmark) equipped with a Pilatus detector.Alkaline stability
The alkaline stability at 90 °C of all six AEMs and at 120 °C of P6Ph-1.8 and P6Ph-2.2 was evaluated after immersion in 2 M aq. NaOH solution for different periods of time. After the immersion, these membranes were washed with deionised water and placed in 1 M aq. NaBr solution to exchange back to the bromide form. After thorough washing and drying, the membranes were dissolved in mixtures of DMSO-d6 and TFA, and analyzed by 1H NMR spectroscopy as described above.Conclusions
A series of copoly(biphenyl alkylene)s carrying N-spirocyclic QA cations was successfully prepared by superacid-mediated polycondensation involving piperidone, biphenyl and trifluoromethylketones, followed by cyclo-quaternisation of the piperidine rings. The copolymers were devoid of diaryl ether linkages and benzylic C–H bonds, and showed very high thermal stability. AEMs based on these copolymers reached high hydroxide ion conductivities at reasonable water contents, balanced by the ionic content of the copolymers. 1H NMR spectroscopy proved crucial to carefully analyse degradation products and identify degradation mechanisms under alkaline conditions. The alkaline stability of the N-spirocyclic QA cations was seemingly limited by distortions of the ring system imposed by the stiff polymer backbone, and hence the ring directly attached to the backbone degraded significantly faster by Hofmann elimination than the pendant ring. Consequently, the results of the present work highlighted the importance of attaching N-spirocyclic QA cations to polymers in such a way that the conformational relaxation of the ring systems is preserved.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Swedish Energy Agency and the Swedish Research Council for financial support. In addition, we are grateful to Peter Holmqvist for assistance with SAXS measurements and data treatment.Notes and references
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7(10), 3135–3191 RSC.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377(1–2), 1–35 CrossRef.
- S. Marini, P. Salvi, P. Nelli, R. Pesenti, M. Villa, M. Berrettoni, G. Zangari and Y. Kiros, Electrochim. Acta, 2012, 82, 384–391 CrossRef.
- M. A. Hickner, A. M. Herring and E. B. Coughlin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51(24), 1727–1735 CrossRef.
- K. D. Kreuer, J. Membr. Sci., 2001, 185(1), 29–39 CrossRef.
- J. A. Kerres, J. Membr. Sci., 2001, 185(1), 3–27 CrossRef.
- A. Kirubakaran, S. Jain and R. K. Nema, Renewable Sustainable Energy Rev., 2009, 13(9), 2430–2440 CrossRef.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K.-i. Kimijima and N. Iwashita, Chem. Rev., 2007, 107(10), 3904–3951 CrossRef PubMed.
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88(4), 981–1007 CrossRef.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38(12), 4901–4934 CrossRef.
- H. Zhang and P. K. Shen, Chem. Rev., 2012, 112(5), 2780–2832 CrossRef PubMed.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y. K. Choe and C. Bae, Macromolecules, 2016, 49(9), 3361–3372 CrossRef.
- T. H. Pham and P. Jannasch, ACS Macro Lett., 2015, 4(12), 1370–1375 CrossRef.
- S. Miyanishi and T. Yamaguchi, Phys. Chem. Chem. Phys., 2016, 18(17), 12009–12023 RSC.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8(3), 513–523 CrossRef PubMed.
- A. D. Mohanty and C. Bae, J. Mater. Chem. A, 2014, 2(41), 17314–17320 RSC.
- C. S. Macomber, J. M. Boncella, B. S. Pivovar and J. A. Rau, J. Therm. Anal. Calorim., 2008, 93(1), 225–229 CrossRef.
- S. A. Nunez and M. A. Hickner, ACS Macro Lett., 2013, 2(1), 49–52 CrossRef.
- J. B. Edson, C. S. Macomber, B. S. Pivovar and J. M. Boncella, J. Membr. Sci., 2012, 399–400, 49–59 CrossRef.
- D. Chen and M. A. Hickner, ACS Appl. Mater. Interfaces, 2012, 4(11), 5775–5781 CrossRef PubMed.
- S. Chempath, B. R. Einsla, L. R. Pratt, C. S. Macomber, J. M. Boncella, J. A. Rau and B. S. Pivovar, J. Phys. Chem. C, 2008, 112(9), 3179–3182 CrossRef.
- O. D. Thomas, K. J. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, J. Am. Chem. Soc., 2012, 134(26), 10753–10756 CrossRef PubMed.
- J. Fan, A. G. Wright, B. Britton, T. Weissbach, T. J. G. Skalski, J. Ward, T. J. Peckham and S. Holdcroft, ACS Macro Lett., 2017, 6(10), 1089–1093 CrossRef.
- K. M. Hugar, H. A. Kostalik and G. W. Coates, J. Am. Chem. Soc., 2015, 137(27), 8730–8737 CrossRef PubMed.
- H.-S. Dang and P. Jannasch, J. Mater. Chem. A, 2016, 4(30), 11924–11938 RSC.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28(2), 1702758 CrossRef.
- H.-S. Dang and P. Jannasch, J. Mater. Chem. A, 2017, 5(41), 21965–21978 RSC.
- F. Gong, R. Wang, X. Chen, P. Chen, Z. An and S. Zhang, Polym. Chem., 2017, 8(29), 4207–4219 RSC.
- Y. Yang, Y. Xu, N. Ye, D. Zhang, J. Yang and R. He, J. Electrochem. Soc., 2018, 165(5), F350–F356 CrossRef.
- J. S. Olsson, T. H. Pham and P. Jannasch, Macromolecules, 2017, 50(7), 2784–2793 CrossRef.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Am. Chem. Soc., 2017, 139(8), 2888–2891 CrossRef PubMed.
- N. Chen, C. Long, Y. Li, C. Lu and H. Zhu, ACS Appl. Mater. Interfaces, 2018, 10(18), 15720–15732 CrossRef PubMed.
- L. Gu, H. Dong, Z. Sun, Y. Li and F. Yan, RSC Adv., 2016, 6(97), 94387–94398 RSC.
- D. J. Strasser, B. J. Graziano and D. M. Knauss, J. Mater. Chem. A, 2017, 5(20), 9627–9640 RSC.
- C. Vogel, H. Komber and J. Meier-Haack, React. Funct. Polym., 2017, 117, 34–42 CrossRef.
- D. A. Klumpp, M. Garza, A. Jones and S. Mendoza, J. Org. Chem., 1999, 64(18), 6702–6705 CrossRef PubMed.
- M. J. O'Connor, K. N. Boblak, A. D. Spitzer, P. A. Gucciardo, A. M. Baumann, J. W. Peter, C. Y. Chen, R. Peter, A. A. Mitton and D. A. Klumpp, Tetrahedron Lett., 2010, 51(38), 4984–4987 CrossRef.
- M. T. Guzmán-Gutiérrez, D. R. Nieto, S. Fomine, S. L. Morales, M. G. Zolotukhin, M. C. G. Hernandez, H. Kricheldorf and E. S. Wilks, Macromolecules, 2011, 44(2), 194–202 CrossRef.
- G. Gebel and O. Diat, Fuel Cells, 2005, 5(2), 261–276 CrossRef.
- J. Pan, S. Lu, Y. Li, A. Huang, L. Zhuang and J. Lu, Adv. Funct. Mater., 2010, 20(2), 312–319 CrossRef.
- W.-H. Lee, Y. S. Kim and C. Bae, ACS Macro Lett., 2015, 4(8), 814–818 CrossRef.
- ASTM D2857-16, Standard Practice for Dilute Solution Viscosity of Polymers, ASTM International, West Conshohocken, PA, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta04699a |
| This journal is © The Royal Society of Chemistry 2018 |